


Integrated transcriptomics identifies ITGB5 as a hub linking coding and non-coding RNAs to signaling regulation in hepatocellular carcinoma
 0
0 Abstract
Aim: To elucidate integrin β5 (ITGB5)-associated transcriptomic alterations in hepatocellular carcinoma cells and identify key coding and non-coding RNA regulators and their associated signaling pathways.
Methods: Whole-transcriptome sequencing was conducted on HepG2 (human hepatocellular carcinoma cell line) cells overexpressing ITGB5. Differential expression analysis was performed, followed by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses for messenger RNAs (mRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs). Candidate transcripts were validated using real-time quantitative polymerase chain reaction (RT-qPCR).
Results: ITGB5 overexpression induced extensive transcriptomic alterations. KEGG enrichment highlighted the phospholipase D, vascular endothelial growth factor (VEGF), and ErbB signaling pathways, with significant involvement of the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) and mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) cascades. Pathway-level analyses suggested a relative attenuation of PI3K/Akt signaling alongside the preferential engagement of MAPK/ERK-related programs. GO analysis indicated shifts in biological processes, notably the negative regulation of peptidase activity and cell-matrix adhesion. Furthermore, ITGB5 was associated with widespread lncRNA and circRNA regulation; predicted target genes of these non-coding RNAs were enriched in PI3K/Akt signaling, protein processing in the endoplasmic reticulum, and cell cycle regulation. RT-qPCR confirmed the upregulation of key transcripts, including dystroglycan 1 (DAG1; mRNA), small nucleolar RNA host gene 1 (SNHG1; lncRNA), and circFN1 (circRNA), in ITGB5-overexpressing cells.
Conclusion: Our findings identify ITGB5 as a regulatory hub in hepatocellular carcinoma (HCC) associated with coding and non-coding RNA regulatory networks, offering insights into potential therapeutic targets.
Keywords
INTRODUCTION
Hepatocellular carcinoma (HCC) remains one of the most prevalent and lethal malignancies worldwide[1,2]. Despite advances in targeted therapies and immunotherapy, the prognosis for patients with advanced HCC remains poor due to high rates of drug resistance, metastasis, and recurrence[3-5]. These challenges underscore the urgent need to identify novel molecular regulators and signaling networks that drive hepatocarcinogenesis and may serve as biomarkers or therapeutic targets.
Integrins play pivotal roles in essential cellular processes - including adhesion, migration, proliferation, and survival - primarily by mediating interactions between cells and the extracellular matrix (ECM) and activating downstream signaling[6]. Aberrant integrin signaling is frequently implicated in tumor invasion, metastatic dissemination, and therapy resistance[7-9]. Among these, integrin β5 (ITGB5) has been shown to promote malignant phenotypes by activating the Wnt/β-catenin (Wingless/Integrated and β-catenin) signaling pathway, transforming growth factor β (TGF-β), and phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) pathways[9,10]. In HCC, elevated ITGB5 expression correlates with aggressive clinicopathological features and poor prognosis[11,12]. However, most studies have focused on protein-level alterations or specific signaling pathways. As a result, the comprehensive transcriptomic programs regulated by ITGB5 in HCC remain largely undefined[7,13].
In the era of precision oncology, integrated transcriptomic analyses incorporating both coding and non-coding RNA layers are essential for elucidating complex regulatory networks. Non-coding RNAs, particularly long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs), have emerged as critical regulators of gene expression in cancer. lncRNAs can influence chromatin dynamics and transcription, while circRNAs frequently function as competing endogenous RNAs (ceRNAs) to modulate target messenger RNA (mRNA) stability[14,15]. Although dysregulation of these non-coding RNAs has been linked to oncogenic pathways in HCC, how ITGB5 integrates these layers into coherent regulatory networks remains poorly understood.
To address these gaps, we performed whole-transcriptome sequencing to compare ITGB5-overexpressing and control HepG2 (human hepatocellular carcinoma cell line) cells. We characterized ITGB5-dependent transcriptomic reprogramming, identified differentially expressed RNAs, and mapped their functional enrichment using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses. Furthermore, we identified dystroglycan 1 (DAG1), small nucleolar RNA host gene 1 (SNHG1), and circFN1 as significant candidates within this network. This work defines a multilayered ITGB5-regulated transcriptomic architecture in HCC and provides a robust framework for future mechanistic investigations into how ITGB5 reshapes oncogenic signaling.
MATERIALS AND METHODS
Cell culture
HepG2 cells were cultured in RPMI-1640 medium (Wisent, Canada) supplemented with 10% fetal bovine serum (FBS; Wisent, China) and 1% penicillin-streptomycin (P/S; Gibco, USA). HEK293T (human embryonic kidney 293T) cells were maintained in high-glucose Dulbecco’s modified Eagle medium (DMEM; Gibco, USA) containing 10% FBS and 1% P/S. Both cell lines were obtained from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and maintained in a humidified incubator at 37 °C with 5% CO2.
Construction of ITGB5 overexpression lentivirus and cell transduction
The coding DNA sequence (CDS) of human ITGB5 [National Center for Biotechnology Information (NCBI) Reference Sequence: NM_002213.5] was amplified from human fibroblast complementary DNA (cDNA) using Q5 High-Fidelity DNA Polymerase (New England Biolabs, USA). The polymerase chain reaction (PCR)-amplified fragment was cloned into the pLV-mCherry vector (Addgene plasmid #36084) via BsrGI and SalI restriction sites. The resulting pLV-mCherry-ITGB5 construct was validated by Sanger sequencing. Lentiviral particles were produced by co-transfecting HEK293T cells with either the pLV-mCherry-ITGB5 or the pLV-mCherry (control) plasmid, along with the packaging vectors HIV-1 Rev expression plasmid (pREV), HIV-1 gag/pol packaging plasmid (pMDL), and vesicular stomatitis virus glycoprotein expression plasmid (pVSVG), using PolyJet transfection reagent (SignaGen Laboratories, USA) according to the manufacturer’s instructions. Viral supernatants were harvested 48 h post-transfection, passed through a 0.45-µm filter, and stored at
HepG2 cells were seeded and cultured to approximately 70% confluence prior to infection. The cells were then transduced with LV-mCherry-ITGB5 or control lentiviral particles in the presence of 5 µg/mL polybrene (Sigma-Aldrich, USA). After a 12 h incubation, the viral suspension was replaced with fresh complete medium, and the cells were maintained for 10 days to establish stable overexpression. ITGB5-overexpressing cells were subsequently harvested for downstream analyses, including RNA extraction and whole-transcriptome sequencing.
RNA extraction and RT-qPCR
Total RNA was extracted from ITGB5-overexpressing and control HepG2 cells using a total
Thermal cycling conditions consisted of an initial denaturation at 95 °C for 30 s, followed by 40 cycles of denaturation at 95 °C for 5 s and annealing/extension at 60 °C for 10 s. Relative gene expression levels were calculated using the 2^-ΔΔCt method. All reactions were performed in triplicate, and each experiment was independently repeated at least three times.
Library preparation and RNA sequencing
Sequencing libraries were constructed using two complementary strategies to profile both coding and non-coding transcriptomes. For mRNA sequencing, 1 µg of total RNA per sample was used to generate libraries with the NEBNext® UltraTM RNA Library Prep Kit for Illumina®. Briefly, poly(A)+ mRNA was enriched using oligo(dT)25 magnetic beads and fragmented. Double-stranded cDNA was synthesized, ligated to single-index adapters, and PCR-amplified.
To comprehensively characterize non-coding RNAs, including lncRNAs, strand-specific libraries were prepared. Ribosomal RNA (rRNA) was depleted using a rRNA Removal Kit, and the remaining RNA was fragmented. Strand-specific cDNA libraries were constructed using a deoxyuridine triphosphate (dUTP)-based second-strand marking strategy, followed by uracil-N-glycosylase digestion to preserve strand orientation. All libraries were assessed for quality using an Agilent Bioanalyzer 2100. Libraries with concentrations > 2 nM were pooled and sequenced on the Illumina NovaSeqTM 6000 platform to generate 150-bp paired-end reads.
Sequence data processing and analysis
Sequence data processing
Raw sequencing reads in FASTQ format were processed with fastp to remove adapter sequences, poly-N reads, and low-quality bases. All libraries exhibited high quality, with Q30 values ranging from 95.79% to 96.74% [Supplementary Table 1]. The resulting clean reads were aligned to the human reference genome (GRCh38) using HISAT2 (v2.0.5), with splice-aware mapping enabled using gene annotation. Transcripts were subsequently assembled using StringTie and merged across samples with gffcompare to generate a unified transcriptome. Gene expression levels were quantified using RNA-Seq by Expectation Maximization (RSEM); specifically, expected read counts were utilized for differential expression analysis, whereas fragments per kilobase of transcript per million mapped reads (FPKM) values were used for expression visualization. Three biological replicates were included for each group.
Differential expression analysis
Differential expression analysis was performed using the DESeq2 R package, which utilizes a negative binomial distribution model to analyze digital gene expression data. To control the false discovery rate (FDR), P values were adjusted for multiple testing using the Benjamini-Hochberg method. Genes meeting the criteria of |log2 fold change| ≥ 2 and an adjusted P value (padj) < 0.05 were considered significantly differentially expressed.
Enrichment analysis of differentially expressed genes
Functional enrichment analyses, including GO and KEGG analyses, were conducted using the clusterProfiler R package to evaluate the statistical enrichment of differentially expressed genes (DEGs). To ensure accuracy, gene length bias was corrected during GO enrichment analysis. Terms and pathways with an adjusted
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was performed to determine whether predefined gene sets showed coordinated differences between the two groups, independent of an explicit DEG cutoff. Briefly, all genes were ranked according to their log2 fold change between the two groups, and predefined gene sets were tested to determine whether their member genes were preferentially enriched at the top or bottom of the ranked list. Gene sets from the GO, KEGG, and Molecular Signatures Database (MSigDB) collections (including Reactome) were analyzed independently. Enrichment significance was assessed using the FDR-adjusted P values, and gene sets with an FDR < 0.05 were considered significantly enriched.
Network analysis
Network analysis was conducted based on significantly enriched KEGG pathways and predicted regulatory relationships. DEGs associated with significantly enriched KEGG pathways were mapped onto KEGG pathway diagrams, and pathway interaction networks were constructed and visualized using Cytoscape. In the pathway network, nodes represent genes or pathways, while edges represent pathway-associated relationships derived from KEGG pathway annotations. For lncRNA-related network analysis, cis- and trans-regulatory networks were generated based on predicted target relationships. Protein-coding genes located within 100 kb upstream or downstream of a given lncRNA were defined as potential cis-target genes[16], while potential trans-target genes were inferred via Pearson correlation analysis between lncRNAs and coding genes[17]. In these networks, nodes represent lncRNAs or target genes, and edges represent predicted regulatory associations. For circRNA-related network analysis, potential microRNA (miRNA) binding sites on differentially expressed circRNAs were predicted using miRanda and TargetScan. Candidate circRNA-miRNA pairs were retained only when they were supported by both tools and satisfied the following thresholds: miRanda score > 140 and free energy < -20 kcal/mol. A circRNA-miRNA interaction network was subsequently constructed based on these associations. In this network, nodes represent circRNAs or miRNAs, and edges represent predicted interaction relationships.
Statistical analysis
All statistical plots were generated using GraphPad Prism 10.0 (GraphPad Software, San Diego, CA, USA). A two-tailed unpaired Student’s t-test was employed for comparisons between two groups. Statistical significance was defined as follows: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
RESULTS
Establishment of the ITGB5 overexpression model and global transcriptomic analysis
To investigate the global regulatory roles of ITGB5, we established a stable ITGB5-overexpressing HepG2 cell line using a lentiviral transduction system. RT-qPCR analysis confirmed a marked increase in ITGB5 mRNA levels in the overexpression group compared with the empty vector control [Supplementary Figure 1].
Transcriptome-wide profiling reveals extensive gene and transcript alterations induced by ITGB5 overexpression
To characterize the transcriptomic impact of ITGB5 overexpression, whole-transcriptome sequencing was performed on ITGB5-OE (ITGB5 overexpression) and control cells. Principal component analysis (PCA) demonstrated a clear separation between the ITGB5-OE and control groups, with biological replicates clustering tightly within each group. These findings confirmed the high reproducibility of our data and the distinct transcriptional profiles induced by ITGB5 overexpression [Figure 1A and B].
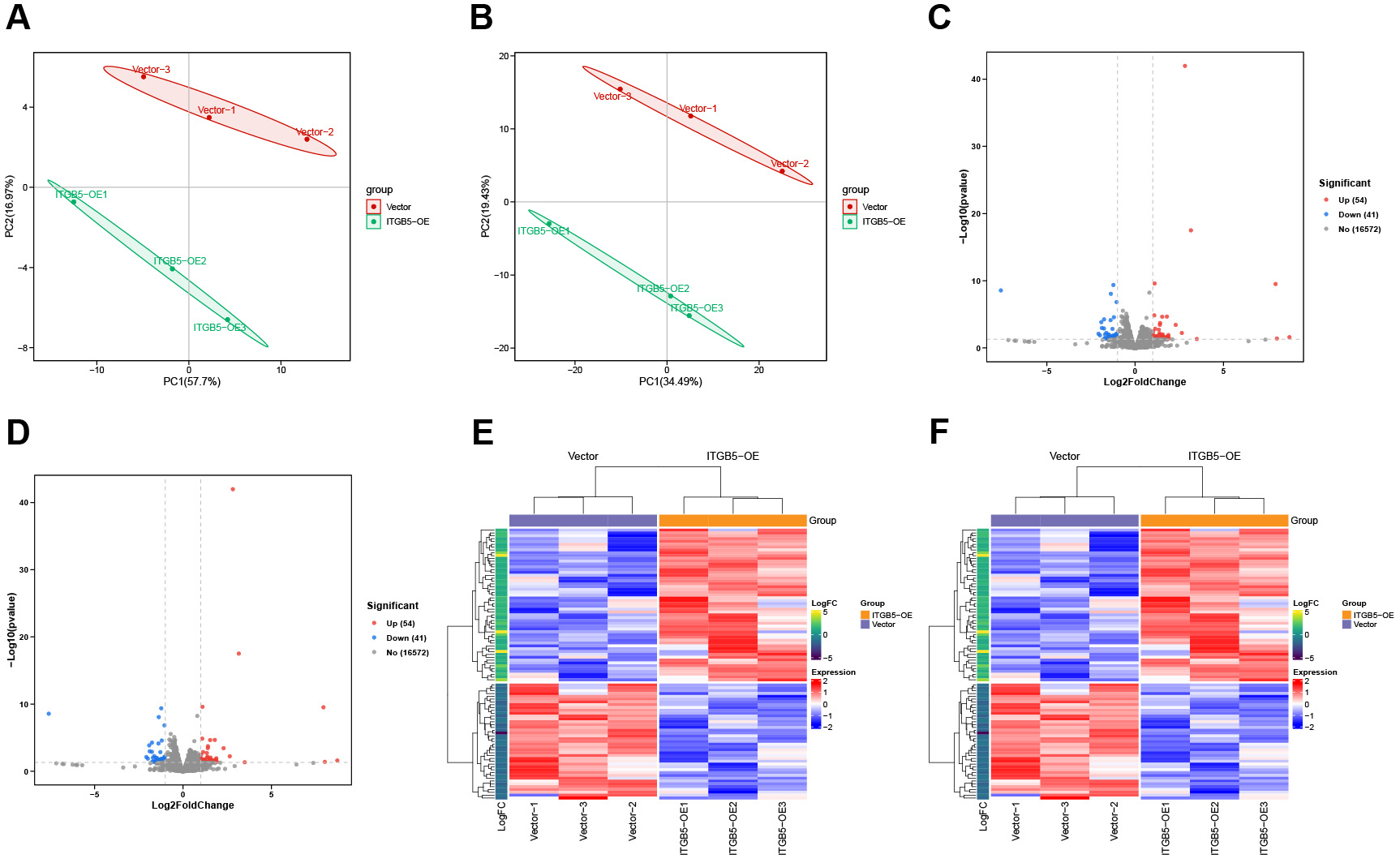
Figure 1. Transcriptomic landscapes and differential expression profiles following ITGB5 overexpression. (A) Principal component analysis (PCA) plot based on differentially expressed genes (DEGs); (B) PCA plot based on differentially expressed transcripts (DETs); (C) Volcano plot depicting DEGs; (D) Volcano plot depicting DETs; (E) Heatmap illustrating the expression of DEGs; (F) Heatmap illustrating the expression of DETs.
DESeq2 analysis identified 95 DEGs (54 upregulated and 41 downregulated) and 2,032 differentially expressed transcripts (DETs; 1,022 upregulated and 1,010 downregulated) [Figure 1C and D, Figure 2A, Supplementary Table 2]. Heatmap visualization further confirmed that these DEGs and DETs were robustly regulated by ITGB5 across all biological replicates [Figure 1E and F].
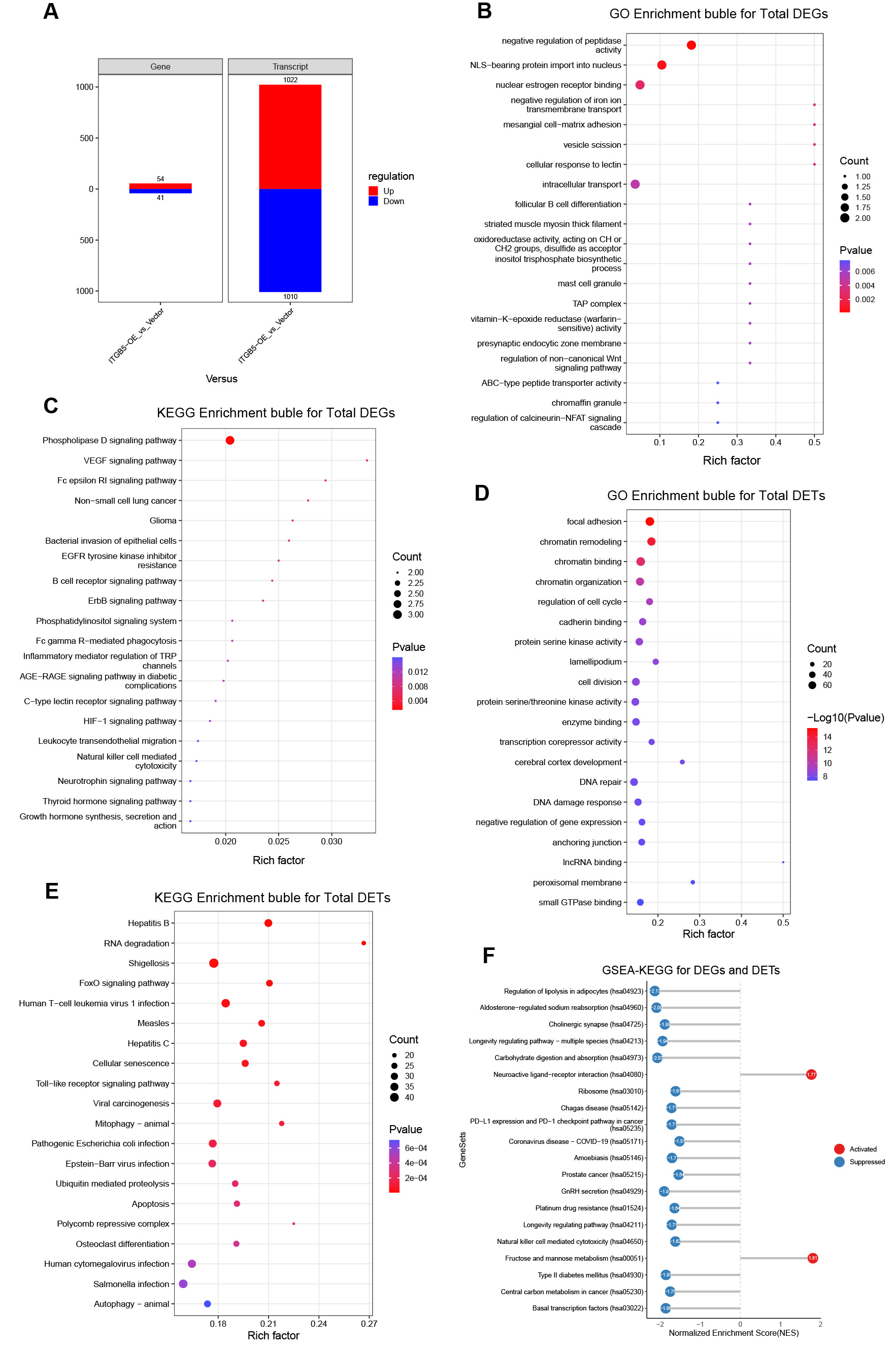
Figure 2. Functional enrichment landscapes of DEGs and DETs following ITGB5 overexpression. (A) Summary of up- and downregulated genes and transcripts; (B) Gene Ontology (GO) enrichment bubble plot for all DEGs; (C) Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment bubble plot for all DEGs; (D) GO enrichment bubble plot for all DETs; (E) KEGG enrichment bubble plot for all DETs; (F) Gene set enrichment analysis based on KEGG pathways (GSEA-KEGG) for integrated DEGs and DETs (activated pathways in red; suppressed pathways in blue). DEGs: Differentially expressed genes; DETs: differentially expressed transcripts; VEGF: vascular endothelial growth factor; TAP: transporter associated with antigen processing; NFAT: nuclear factor of activated T cells; NLS: nuclear localization signal; IRF: interferon regulatory factor; EGFR: epidermal growth factor receptor; TRP: transient receptor potential; AGE-RAGE: advanced glycation end products-receptor for advanced glycation end products; HIF-1: hypoxia-inducible factor 1; IncRNA: long non-coding RNA.
ITGB5 broadly activates oncogenic signaling pathways and modulates key cellular functions
To examine the biological implications of ITGB5-driven transcriptional changes, we conducted GO and KEGG enrichment analyses on the identified DEGs. Overall, ITGB5 overexpression was predominantly associated with pathways related to cell adhesion, oncogenic signaling, and intracellular regulatory processes.
At the gene level, GO enrichment analysis highlighted biological programs linked to cell-matrix adhesion and the regulation of proteolysis [Figure 2B and Supplementary Table 3]. Concurrently, KEGG analysis indicated that the DEGs were primarily enriched in pathways converging on the PI3K/Akt and mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling axes, including phospholipase D, vascular endothelial growth factor (VEGF), and ErbB signaling [Figure 2C and Supplementary Table 4].
At the transcript level, GO enrichment of DETs revealed processes associated with cell architecture and epigenetic regulation, including focal adhesion, chromatin remodeling, and serine/threonine kinase activity [Figure 2D and Supplementary Table 5]. KEGG analysis of these DETs further indicated enrichment in pathways related to viral infections (including hepatitis B and C), cellular senescence, and FoxO signaling [Figure 2E and Supplementary Table 6].
Convergence of complex signaling networks on PI3K/Akt and MAPK/ERK pathways
To further characterize the signaling architecture downstream of ITGB5, we performed complementary GSEA and network analyses. GSEA using GO and MSigDB/Reactome gene sets revealed a clear
Network analysis of significantly enriched KEGG pathways highlighted PI3K/Akt and MAPK/ERK as major convergent nodes, supporting their roles as core regulatory axes in the ITGB5 downstream network [Supplementary Figure 2C]. Furthermore, KEGG-GSEA confirmed the perturbation of these two cascades across multiple enriched pathways, including the cholinergic synapse, longevity-regulating pathways, carbohydrate digestion and absorption, the programmed death ligand 1/programmed cell death protein 1 (PD-L1/PD-1) checkpoint, prostate cancer, gonadotropin-releasing hormone (GnRH) secretion, platinum drug resistance, natural killer (NK) cell-mediated cytotoxicity, and central carbon metabolism in cancer [Figure 2F].
Functional enrichment of up- and downregulated DEGs
Differential expression analysis further revealed distinct functional signatures between upregulated and downregulated DEGs. Specifically, upregulated genes were primarily enriched in signaling and membrane-trafficking programs associated with the phospholipase D, VEGF, and ErbB-related pathways, consistent with the activation of ITGB5-linked oncogenic signaling [Figure 3A and B]. In contrast, downregulated genes were predominantly associated with antigen presentation, peptidase regulation, and selected metabolic or endocrine-related pathways [Figure 3C and D]. Collectively, these findings suggest that ITGB5 overexpression preferentially amplifies signaling programs governing cell communication and tumor progression while suppressing a subset of homeostatic and regulatory processes.
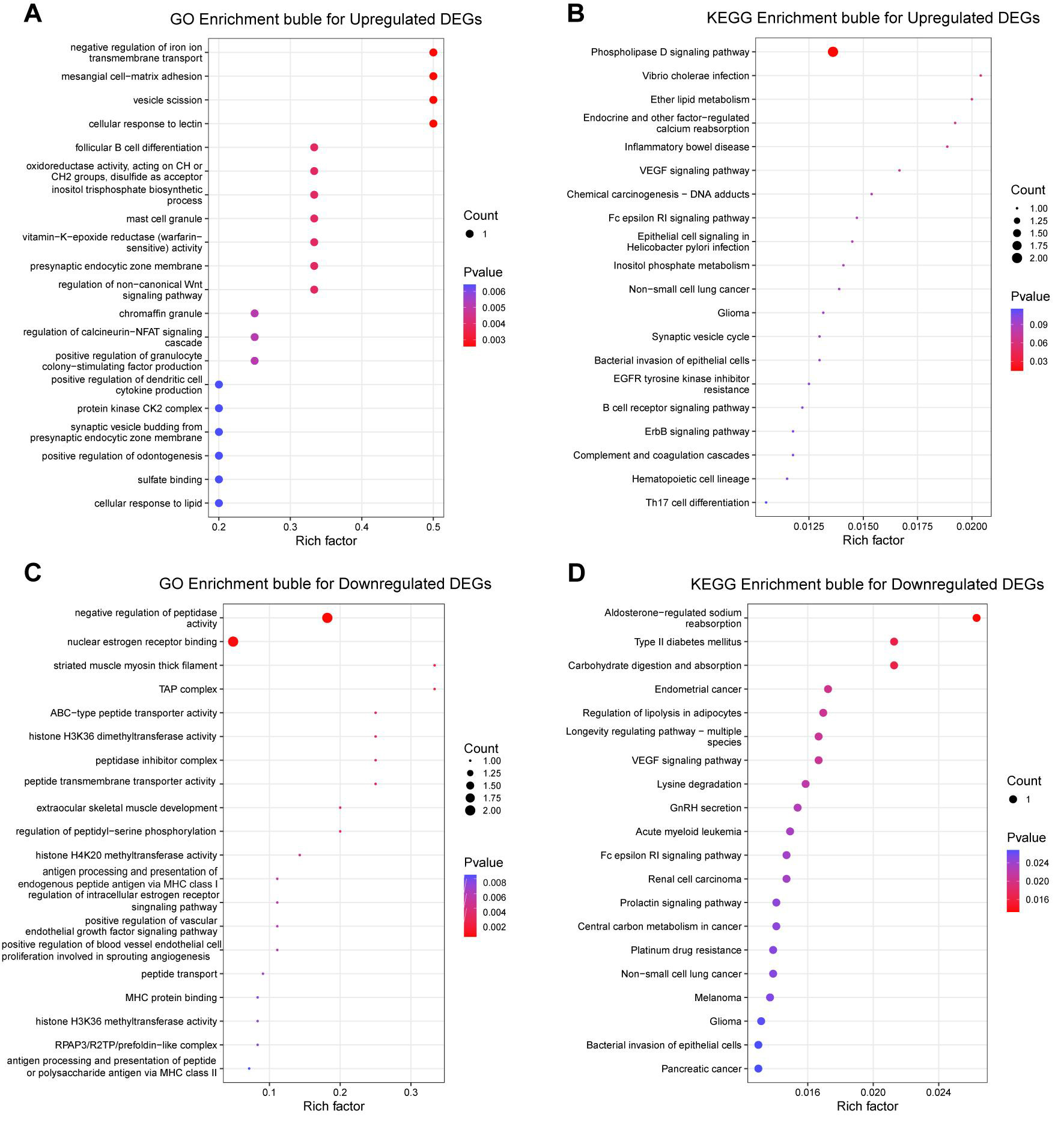
Figure 3. GO and KEGG enrichment analyses of upregulated and downregulated DEGs following ITGB5 overexpression. (A) GO enrichment bubble plot for upregulated DEGs; (B) KEGG enrichment bubble plot for upregulated DEGs; (C) GO enrichment bubble plot for downregulated DEGs; (D) KEGG enrichment bubble plot for downregulated DEGs. DEGs: Differentially expressed genes; DETs: differentially expressed transcripts; GO: gene ontology; KEGG: kyoto encyclopedia of genes and genomes; VEGF: vascular endothelial growth factor; EGFR: epidermal growth factor receptor; TAP: transporter associated with antigen processing; ABC: ATP-binding cassette; MHC: major histocompatibility complex; NFAT: nuclear factor of activated T cells.
Functional enrichment of up- and downregulated DETs
GO and KEGG analyses of DETs demonstrated that ITGB5-induced transcript-level alterations were primarily linked to cell-matrix interaction, intracellular trafficking, chromatin regulation, and RNA processing. Upregulated transcripts were enriched in focal adhesion- and signaling-related processes
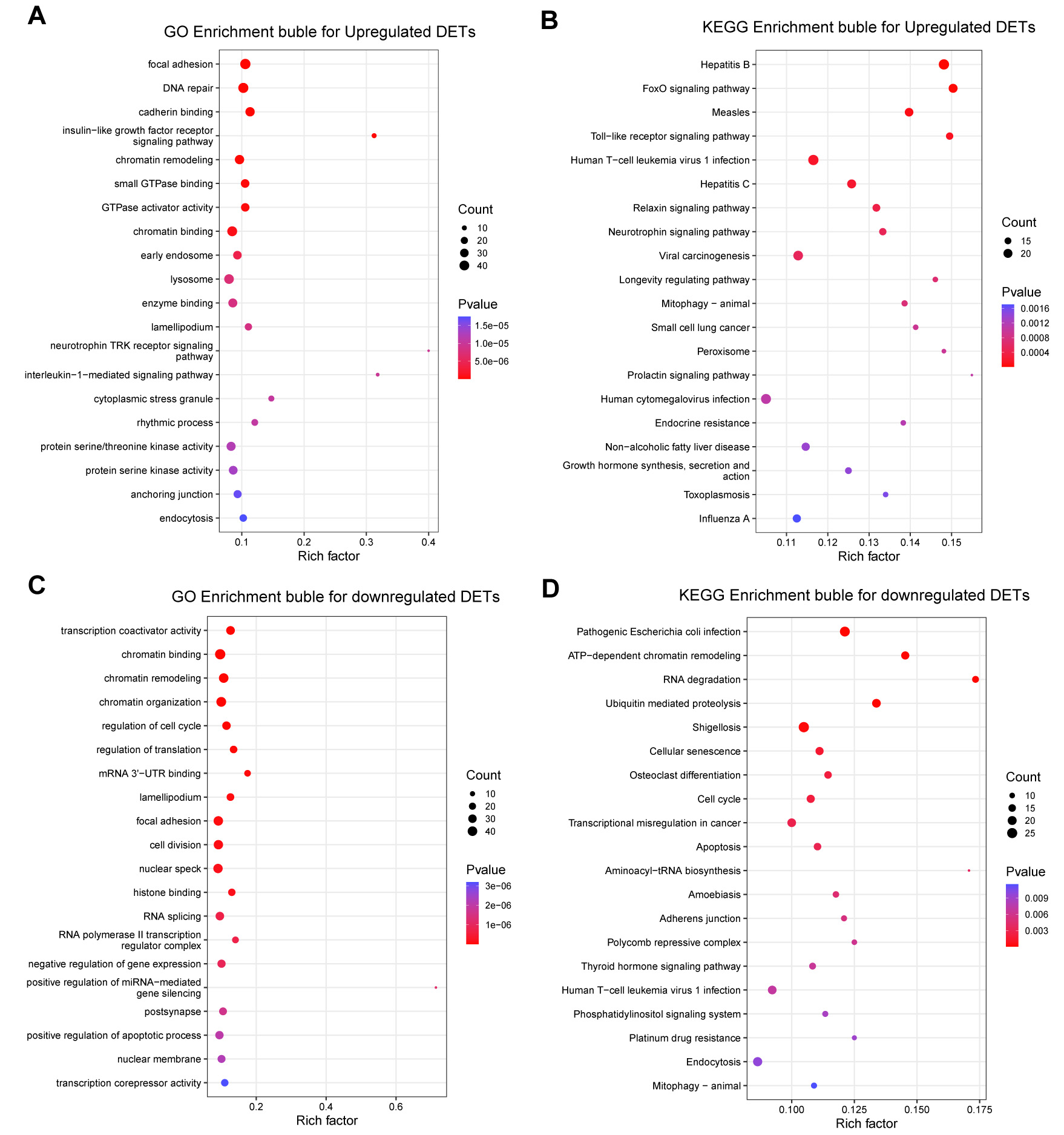
Figure 4. GO and KEGG enrichment profiles of upregulated and downregulated DETs following ITGB5 overexpression. (A) GO enrichment bubble plot illustrating upregulated DETs; (B) KEGG enrichment bubble plot illustrating upregulated DETs; (C) GO enrichment bubble plot illustrating downregulated DETs; (D) KEGG enrichment bubble plot illustrating downregulated DETs. DEGs: Differentially expressed genes; DETs: differentially expressed transcripts; GO: gene ontology; KEGG: kyoto encyclopedia of genes and genomes; TRK: tropomyosin receptor kinase; 3′UTR: 3′ untranslated region; ATP: adenosine triphosphate.
ITGB5 regulates a non-coding RNA network associated with stress response and signal transduction
To further elucidate the regulatory effects of ITGB5 beyond protein-coding genes, we examined its impact on non-coding RNAs. At the transcript level, we identified 89,832 mRNAs alongside 223,395 annotated lncRNA transcripts and 1,142 novel lncRNA candidates [Supplementary Figure 3A], which were broadly distributed across the genome [Supplementary Figure 3B]. Differential analysis revealed 385 lncRNA transcripts significantly altered by ITGB5 overexpression (174 upregulated and 211 downregulated), as illustrated by volcano plot and hierarchical clustering analyses [Supplementary Figure 3C and D, Supplementary Table 7]. When summarized at the gene level, 39 non-coding RNA loci were differentially expressed (17 upregulated and 22 downregulated), and these changes similarly distinguished ITGB5-OE from vector controls [Supplementary Figure 3E and F, Supplementary Table 8].
To explore the functional implications of ITGB5-regulated lncRNAs, we predicted their cis- and trans-target genes and visualized the corresponding regulatory networks [Figure 5A and B, Supplementary Table 9]. GO enrichment of cis-target genes indicated associations with cellular responses to hypoxia, RNA splicing/mRNA degradation, protein quality control, and integrin-mediated signaling [Figure 5C]. Concurrently, KEGG analysis highlighted significant enrichment in the PI3K/Akt signaling pathway and protein processing in the endoplasmic reticulum [Figure 5D]. In parallel, GO enrichment of trans-target genes was dominated by chromatin remodeling, endoplasmic reticulum stress, RNA splicing, and the regulation of apoptosis [Figure 5E], while KEGG analysis further supported enrichment in RNA splicing-related pathways and the endoplasmic reticulum unfolded protein response (UPR) [Figure 5F].
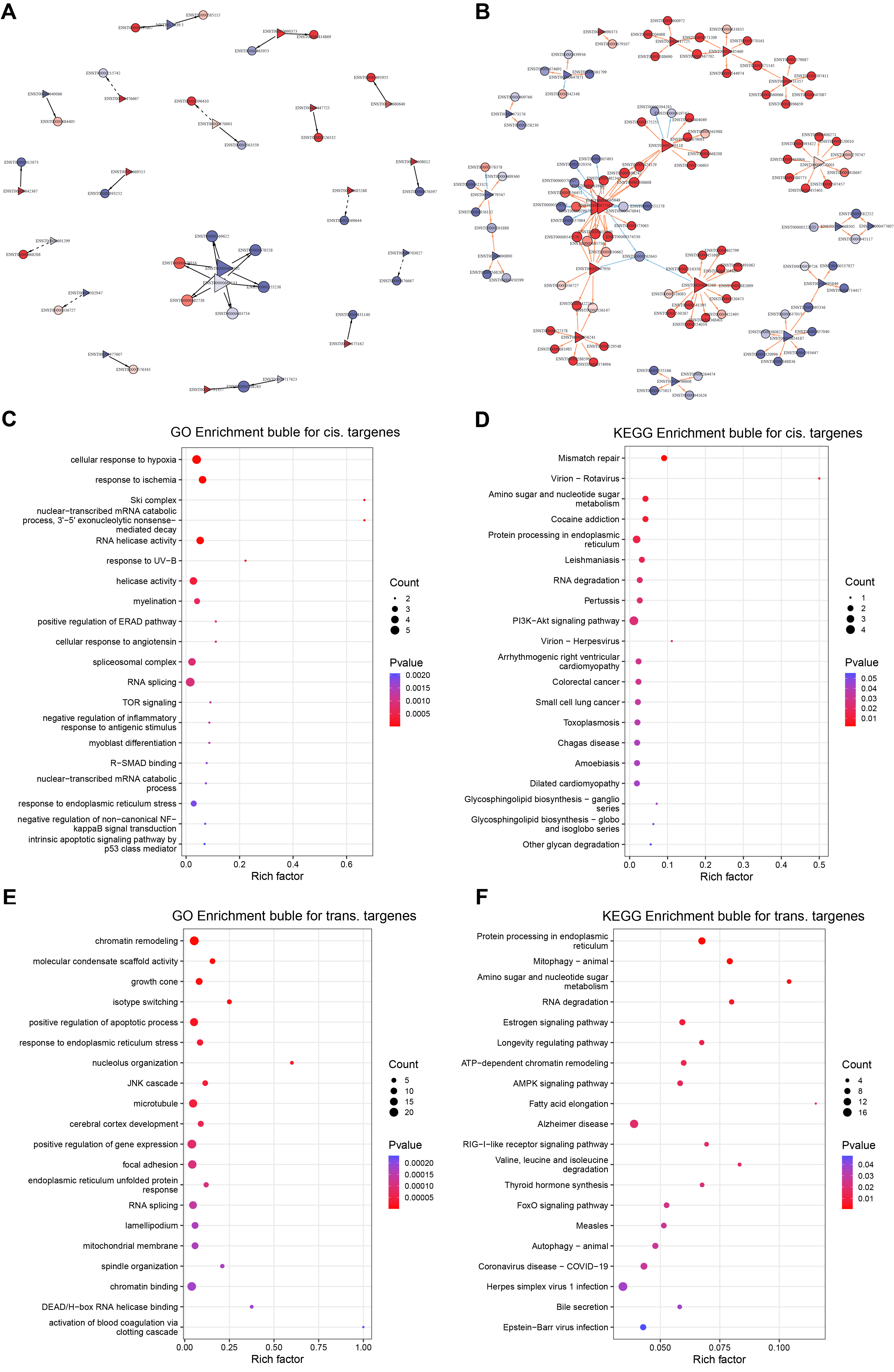
Figure 5. Predicted long non-coding RNAs (lncRNA) targets and functional enrichment profiles. (A) Cis-target regulatory network of ITGB5-responsive lncRNAs; (B) Trans-target regulatory network of ITGB5-responsive lncRNAs; (C) GO enrichment bubble plot depicting cis-target genes; (D) KEGG enrichment bubble plot depicting cis-target genes; (E) GO enrichment bubble plot depicting trans-target genes; (F) KEGG enrichment bubble plot depicting trans-target genes. DEGs: Differentially expressed genes; DETs: differentially expressed transcripts; GO: gene ontology; KEGG: kyoto encyclopedia of genes and genomes; PI3K/Akt: phosphoinositide 3-kinase/protein kinase B; UV-B: ultraviolet B; ERAD: endoplasmic reticulum-associated degradation; TOR: target of rapamycin; R-SMAD: receptor-regulated SMAD; NF: nuclear factor; JNK: c-Jun N-terminal kinase; ATP: adenosine triphosphate; AMPK: AMP-activated protein kinase; RIG: retinoic acid-inducible gene.
Consistent with these transcriptome-wide changes, circRNA profiling indicated that ITGB5 overexpression modulated the circRNA landscape without markedly altering global size characteristics, as circRNA length distributions were highly similar between groups [Figure 6A]. Sample-wise expression patterns further confirmed robust circRNA detection across replicates [Figure 6B]. Differential analysis identified 251 dysregulated circRNAs (120 upregulated and 131 downregulated) in ITGB5-OE versus vector controls [Figure 6C and Supplementary Table 10]. Heatmap visualization coupled with unsupervised hierarchical clustering clearly distinguished ITGB5-OE samples from controls [Figure 6D], indicating a distinct ITGB5-associated circRNA signature.
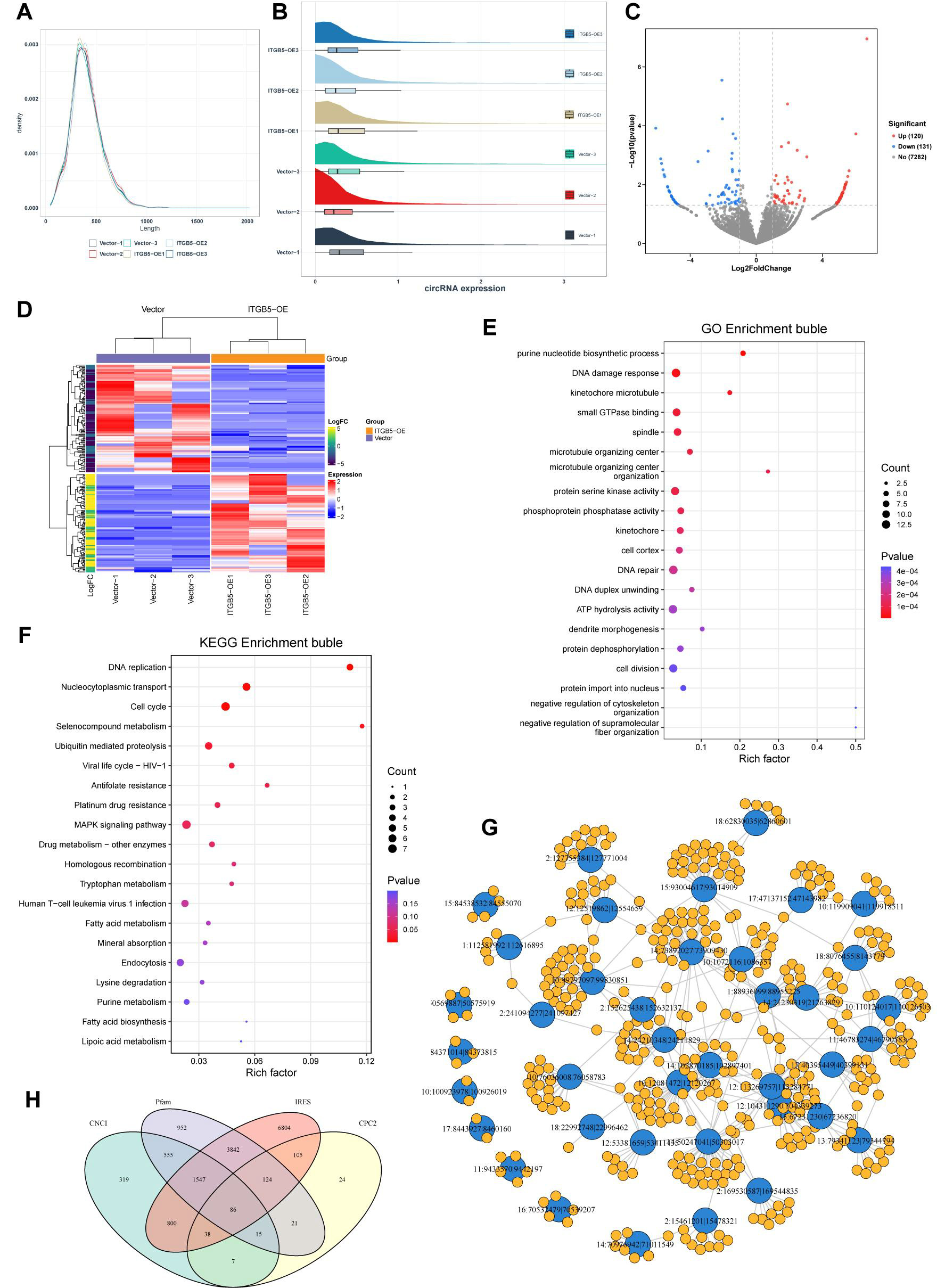
Figure 6. Characterization of circular RNAs (circRNAs) and functional enrichment analysis of their host genes. (A) Length distribution of identified circRNAs; (B) Box plot illustrating the distribution of circRNA expression across samples; (C) Volcano plot depicting differentially expressed circRNAs; (D) Heatmap showing differentially expressed circRNAs with unsupervised hierarchical clustering; (E) GO enrichment bubble plot for the host genes of dysregulated circRNAs; (F) KEGG enrichment bubble plot of the host genes for dysregulated circRNAs; (G) Predicted circRNA-miRNA interaction network; (H) Venn diagram illustrating circRNAs with predicted coding potential based on four algorithms: CNCI, Pfam, IRES, and CPC2. GO: Gene ontology; KEGG: kyoto encyclopedia of genes and genomes; CNCI: coding-non-coding index; IRES: internal ribosome entry site; CPC2: coding potential calculator; ATP: adenosine triphosphate; MAPK: mitogen-activated protein kinase.
To infer potential functions, we analyzed the host genes of the dysregulated circRNAs. GO enrichment was dominated by genome integrity and mitotic regulation, including the DNA damage response/repair, microtubule-kinetochore organization, and cell division-related processes [Figure 6E]. KEGG analysis reinforced these themes, highlighting DNA replication, the cell cycle, homologous recombination, and ubiquitin-mediated proteolysis, as well as nucleocytoplasmic transport and MAPK signaling [Figure 6F]. In parallel, the predicted circRNA-miRNA interaction network suggested extensive regulatory connectivity [Figure 6G], consistent with a potential post-transcriptional modulatory role for ITGB5-responsive circRNAs. Finally, coding-potential assessment showed minimal overlap among predictions from Coding-Non-Coding Index (CNCI), Coding Potential Calculator 2 (CPC2), Pfam, and internal ribosome entry site (IRES) [Figure 6H], supporting the conclusion that most identified circRNAs are unlikely to encode peptides.
Validation of candidate genes by RT-qPCR
To further validate the ITGB5-associated multilayered regulatory landscape suggested by our enrichment and network analyses, we selected three representative DETs for RT-qPCR validation: DAG1 (mRNA; encoding dystroglycan 1, an ECM receptor), SNHG1 (lncRNA; a reported oncogenic lncRNA), and circFN1 (a circRNA derived from the FN1 gene). These candidates were prioritized because they were integral to our pathway-focused analyses linked to the PI3K/Akt and MAPK/ERK signaling axes. RT-qPCR confirmed that the expression levels of all three transcripts were significantly upregulated in ITGB5-overexpressing cells compared with the vector controls [Figure 7A-C], consistent with the RNA-seq results. Detailed results are presented in Supplementary Table 11.
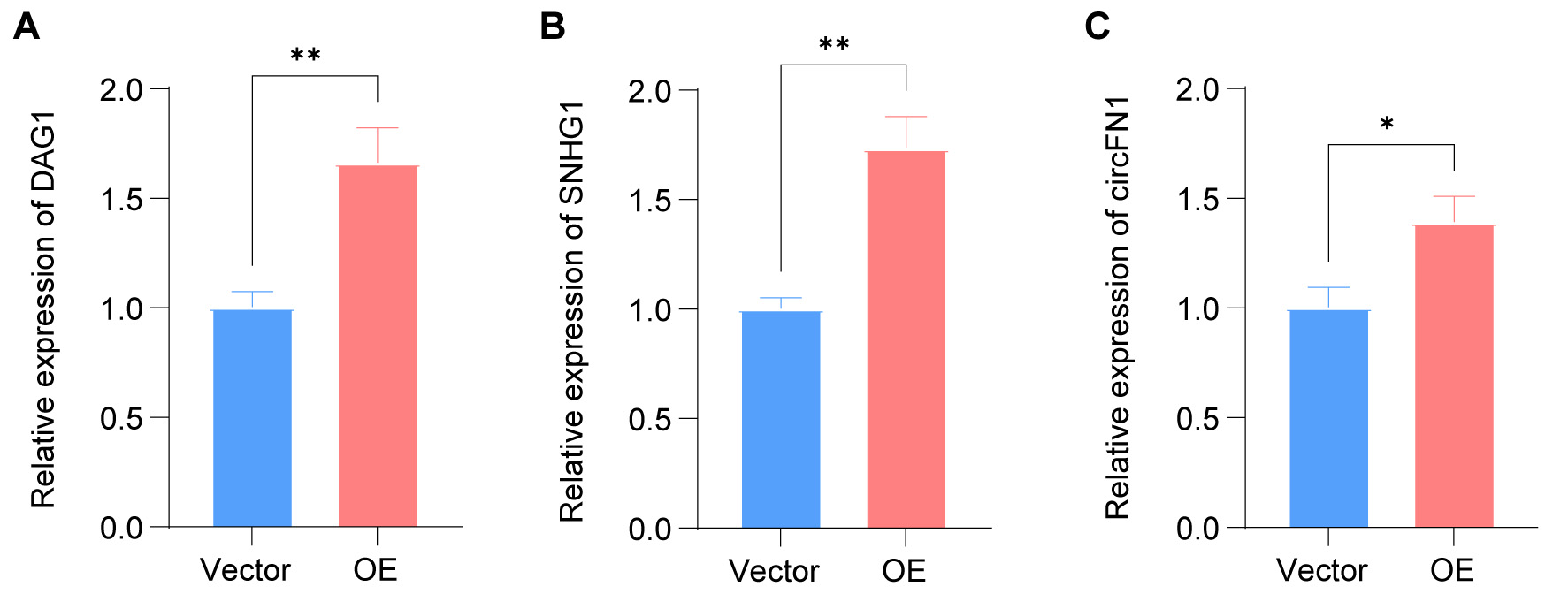
Figure 7. Validation of key regulatory nodes following ITGB5 overexpression by RT-qPCR. The relative expression levels of three selected candidate molecules were assessed: (A) DAG1 mRNA, (B) SNHG1 lncRNA, and (C) circFN1. Data are presented as mean ± SD (n = 3). Statistical significance was determined using an unpaired two-tailed Student’s t-test. *P < 0.05, **P < 0.01. RT-qPCR: Real-time quantitative polymerase chain reaction; mRNA: messenger RNA; lncRNA: long non-coding RNAs; OE: overexpression; SD: standard deviation.
DISCUSSION
HCC remains a leading cause of cancer-related mortality worldwide, with advanced-stage disease exhibiting a particularly poor prognosis due to limited effective therapeutic options[18,19]. The pronounced molecular heterogeneity of HCC continues to impede the development of broadly effective targeted therapies, underscoring the urgent need to elucidate the novel regulatory networks that drive hepatocarcinogenesis[20-22]. In this context, our integrated transcriptomic analyses identify ITGB5 as a candidate regulatory hub associated with complex coding and non-coding RNA networks. These findings provide a systems-level perspective on ITGB5-driven regulatory mechanisms and their specific roles in HCC pathobiology.
The high mortality associated with HCC is largely attributable to its aggressive clinical behavior and intrinsic resistance to conventional treatments[4,23]. Increasing evidence suggests that dysregulated signaling networks and complex tumor-microenvironment interactions play pivotal roles in driving HCC progression[24,25]. Within this landscape, integrins have emerged as key coordinators of ECM communication, integrating biochemical and mechanical cues to modulate cell adhesion, migration, proliferation, and survival. ITGB5, in particular, has been linked to invasive growth and metastatic dissemination across multiple malignancies[9,26]. Despite these advances, the global downstream transcriptomic landscape governed by ITGB5 in HCC - particularly concerning lncRNAs and circRNAs - remains largely unexplored. Our study addresses this knowledge gap by characterizing ITGB5-regulated coding and non-coding RNA networks and demonstrating their convergence on core oncogenic signaling pathways.
Paradoxically, our observation that PI3K/Akt signaling is consistently suppressed following ITGB5 overexpression contrasts with the widely recognized role of ITGB5 in promoting PI3K/Akt activation[27,28]. However, this finding aligns with accumulating evidence that integrin-mediated signaling is governed by intricate, often nonlinear regulatory architectures. Prior studies have demonstrated that integrin engagement can activate a PI3K-ARAP3 (ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 3)-dependent negative feedback loop that locally attenuates PI3K activity[29], offering a plausible mechanistic basis for the suppression observed in our model. Moreover, extensive crosstalk among the FAK/Src, PI3K/Akt, and MAPK pathways has been well documented, and recent reviews emphasize the profound context dependence of integrin signaling and its ability to reprogram downstream outputs under distinct cellular states[30]. Supporting this context specificity, the integrin-PI3K/Akt axis exhibits cell type-dependent signaling patterns in HCC models, including HepG2 cells[31]. Computational systems biology analyses further reinforce this view by demonstrating that relative protein abundance and signaling stoichiometry critically shape pathway prioritization and output specificity[32]. Additionally, the suppression of PI3K/Akt signaling may reflect an adaptive response to oncogenic stress, wherein cancer cells activate compensatory programs, such as stress-response pathways, autophagy, and metabolic rewiring, to maintain cellular homeostasis under persistent oncogenic pressure[33,34]. Together, these literature-based considerations may explain the paradoxical PI3K/Akt suppression observed in our study; however, these mechanisms were not directly tested here and should therefore be interpreted cautiously.
Our data suggest that ITGB5 is associated with alterations in the expression of both lncRNAs and circRNAs, thereby influencing key cellular processes, including PI3K/Akt signaling, endoplasmic reticulum stress responses, and cell cycle regulation. Notably, ITGB5 overexpression upregulated DAG1, SNHG1, and circFN1 - molecules intimately linked to PI3K/Akt and MAPK/ERK signaling - yet was associated with the suppression of PI3K/Akt signaling and the preferential activation of MAPK/ERK in HepG2 cells. This non-canonical pattern highlights the context-dependent nature of ITGB5-mediated signaling and suggests the presence of finely tuned feedback and compensatory mechanisms.
A representative example of this context-dependence is DAG1. Although the dystroglycan complex is generally associated with PI3K/Akt activation, our findings indicate that PI3K/Akt signaling is suppressed in ITGB5-overexpressing cells despite DAG1 upregulation. This apparent discrepancy may reflect the functional duality of DAG1, which can exert either activating or inhibitory effects depending on the cellular context. In our system, ITGB5-associated upregulation of DAG1 may be linked to context-specific inhibitory effects. Furthermore, the intracellular domain of dystroglycan has been reported to facilitate ERK activation by recruiting MEK/ERK components to the plasma membrane[35], a mechanism that likely contributes to the enhanced MAPK/ERK signaling observed in our HCC model.
Recent studies have highlighted the multifaceted roles of non-coding RNAs, including lncRNAs, circRNAs, and other small RNA species, in oncogenic regulation. In this context, lncRNAs are known to modulate cancer-related biological processes such as migration, invasion, proliferation, and apoptosis, while circRNAs often function as miRNA sponges or protein regulators, thereby contributing to tumor progression through various signaling pathways[36,37]. These observations further support the biological relevance of the lncRNA- and circRNA-associated regulatory landscapes identified in the present study. Among these, SNHG1 has been reported to promote tumorigenesis via ceRNA-mediated sequestration of tumor-suppressive miRNAs, such as miR-195, thereby activating the PI3K/Akt/mechanistic target of rapamycin (mTOR) signaling axis[38-40]. Intriguingly, in ITGB5-overexpressing HepG2 cells, SNHG1 upregulation was accompanied by suppression of the PI3K/Akt pathway and activation of the MAPK/ERK signaling. This finding suggests a potential reprogramming of signaling hierarchies under ITGB5-high conditions, although the underlying ceRNA mechanisms remain to be fully elucidated. Given that ITGB5 predominantly signals through the focal adhesion kinase (FAK)/Src/RAS (rat sarcoma)/ERK cascade, this route may become the dominant signaling output under ITGB5-high conditions. Consequently, PI3K/Akt suppression may emerge from robust negative feedback or enhanced phosphatase activity [e.g., phosphatase and tensin homolog (PTEN) or SH2-containing inositol phosphatase (SHIP)][41,42].
Similarly, PI3K/Akt suppression may involve the circFN1-miR-182 regulatory axis. Although circFN1 has been reported to function as a competing ceRNA that sponges miR-182, the downstream effects of miR-182 vary significantly across cancer types[43,44]. In our model, circFN1-associated regulation may be linked to PI3K/Akt inhibition, suggesting a context-dependent outcome. Furthermore, circFN1 has been documented to interact with miR-218, thereby influencing MAPK signaling[45] and potentially contributing to pathway crosstalk Supplementary Figure 4. However, these interactions were not experimentally validated in the present study and warrant further investigation.
Through targeted RT-qPCR validation, we confirmed the dysregulation of representative RNA regulators, including DAG1, SNHG1, and circFN1, thereby supporting the robustness of our transcriptomic findings. The high concordance between our sequencing data and RT-qPCR results strengthens the reliability of the ITGB5-associated regulatory network identified in this study.
The translational relevance of our findings is noteworthy. Recent studies have emphasized that integrated biomarker strategies are increasingly pivotal for cancer classification and personalized management[46]. In this context, the ITGB5-associated RNA network identified herein may provide a preliminary framework for RNA-based biomarker development and molecular stratification in HCC. In addition, the growing interest in utilizing non-coding RNAs as diagnostic and prognostic tools[47] further supports the clinical relevance of these findings. However, these implications remain preliminary and require rigorous validation in independent clinical cohorts and through further functional studies.
Nonetheless, several limitations of this study should be noted. First, our findings are primarily based on in vitro cellular models, which, although suitable for mechanistic exploration, require further validation in animal models and patient-derived tissues to establish their physiological and clinical relevance. Furthermore, all experiments were conducted exclusively in HepG2 cells. While HepG2 is a widely utilized HCC model, validation in additional cell lines - such as Huh7 or LM3 (human hepatocellular carcinoma cell lines) - is necessary to confirm the broader applicability of our findings. Second, the inferred modulation of key signaling pathways, particularly PI3K/Akt and MAPK/ERK, is based predominantly on transcriptomic data. As RNA-level changes do not necessarily reflect protein abundance or activation status - especially for signaling molecules regulated by phosphorylation - protein-level validation of representative components, such as the phosphorylation status of AKT and ERK, would provide essential complementary evidence. Accordingly, future studies will prioritize such experiments to further substantiate the role of ITGB in regulating signaling mechanisms. Third, although we have delineated an ITGB5-associated transcriptomic network, the precise regulatory relationships among its components remain to be fully elucidated. Crucially, because ITGB5 knockdown or knockout experiments were not performed, a direct causal relationship between ITGB5 and the observed transcriptomic alterations cannot be established. Similarly, while DAG1, SNHG1, and circFN1 were identified as ITGB5-associated candidates, their direct or indirect regulation by ITGB5, as well as the underlying mechanisms - including transcriptional control and potential competing ceRNA interactions - require rigorous experimental validation. Additionally, integrin-mediated signaling is inherently complex and involves extensive crosstalk among different integrin subunits. ITGB5 forms heterodimers with specific α subunits, and alterations in its expression may influence the composition and balance of integrin complexes at the cell surface[48]. Moreover, different integrin complexes can converge on shared signaling nodes, including the FAK/Src, PI3K/Akt, and MAPK/ERK pathways, enabling cooperative or competitive regulation of downstream signaling outputs[49]. In HCC, such network-level interactions are plausible, as other integrin modules have also been linked to AKT/mTOR and FAK-associated signaling[31]. Therefore, the signaling effects observed in this study may be influenced, at least in part, by interactions with other integrin subunits; however, these crosstalk mechanisms were not directly investigated and warrant further evaluation. Finally, the lack of validation in clinical HCC samples represents an additional limitation. Future studies should incorporate clinical specimens to investigate the clinical associations between ITGB5, its downstream RNA candidates, and specific clinicopathological features or patient outcomes.
Moving forward, further work is needed to clarify how ITGB5 regulates DAG1, SNHG1, and circFN1 at the transcriptional or post-transcriptional levels, to define their functional roles through gain- and loss-of-function assays, and to determine how these effectors collectively influence the balance between PI3K/Akt and MAPK/ERK signaling. The integration of these regulatory mechanisms within the ITGB5-driven oncogenic network remains a key area for future investigation.
Conclusion
In conclusion, our integrated transcriptomic profiling indicates that ITGB5 overexpression is associated with coordinated alterations of coding and non-coding RNAs in HCC, exemplified by DAG1, SNHG1, and circFN1, and with prominent involvement of the PI3K/Akt and MAPK/ERK signaling axes. These results expand our understanding of ITGB5-linked transcriptional reprogramming in HCC and identify ITGB5 and selected downstream RNA effectors as candidate biomarkers and potential therapeutic targets for further validation in clinically relevant models.
DECLARATIONS
Acknowledgments
The Graphical Abstract was created with BioRender.com and is available at: https://app.biorender.com/illustrations/6980711e720b70dc99a86a98.
Authors’ contributions
Made substantial contributions to conception and design of the study: Yang, D.; Chen, X.
Developed the methodology and performed the investigation: Shen, W.; Wang, J.; Zeng, Q.
Performed formal analysis and interpreted the data: Xin, Q.; Lin, Z.; Li, G.
Performed data curation and drafted the original manuscript: Shen, W.
Provided project administration, supervision, and funding acquisition: He, J.; Yang, D.
Revised and edited the manuscript: He, J.; Yang, D.; Chen, X.
All authors read and approved the submitted version.
Availability of data and materials
RT-qPCR source data are provided in the Supplementary Materials. RNA-seq raw data are being prepared for submission to the Sequence Read Archive (SRA) and will be made publicly available upon completion of the deposition process.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by grants from Noncommunicable Chronic Diseases-National Science and Technology Major Project (2024ZD0531200); National Natural Science Foundation of China (82172205, 82402915, 82302842, 82560451); Guangdong Basic and Applied Basic Research Foundation (2023A1515140123, 2024A1515220094, 2024A1515140053, 2025A1515012214); The Medical Clinical Key Specialty Construction Project for 14th Five-Year Plan of Guangdong Province; The Medical High Level-Key Specialty Construction Project for 14th Five-Year Plan of Foshan City; Foshan Key Laboratory of Tissue Engineering for Skin and Precision Diagnosis and Treatment of Infectious Skin Diseases; National Key Clinical Specialty Construction Program (Department of Burns, Plastic Surgery and Wound Repair).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229-63.
2. Singal AG, Kanwal F, Llovet JM. Global trends in hepatocellular carcinoma epidemiology: implications for screening, prevention and therapy. Nat Rev Clin Oncol. 2023;20:864-84.
3. Luo Y, Zhu H. Immunotherapy for advanced or recurrent hepatocellular carcinoma. World J Gastrointest Oncol. 2023;15:405-24.
4. Zou Y, Wan X, Zhou Q, et al. Mechanisms of drug resistance in hepatocellular carcinoma. Biol Proced Online. 2025;27:19.
5. Lee YR. A multidisciplinary approach with immunotherapies for advanced hepatocellular carcinoma. J Liver Cancer. 2023;23:316-29.
6. Yuan Z, Yan K, Wang J. Overexpression of integrin β2 improves migration and engraftment of adipose-derived stem cells and augments angiogenesis in myocardial infarction. Ann Transl Med. 2022;10:863.
7. Zhang L, Guo Q, Guan G, Cheng W, Cheng P, Wu A. Integrin beta 5 is a prognostic biomarker and potential therapeutic target in glioblastoma. Front. Oncol. 2019;9:904.
8. Shi W, He J, Huang Y, et al. Integrin β5 enhances the malignancy of human colorectal cancer by increasing the TGF-β signaling. Anticancer Drugs. 2021;32:717-26.
9. Cheng Y, Lin X, Xu H, et al. Integrin β5, a noninvasive diagnostic biomarker, is associated with unfavorable prognosis and immunotherapy efficacy in gastric cancer. BMC Gastroenterol. 2024;24:362.
10. Chen Z, Fang Y, Zhong S, Lin S, Yang X, Chen S. ITGB5 is a prognostic factor in colorectal cancer and promotes cancer progression and metastasis through the Wnt signaling pathway. Sci Rep. 2025;15:9225.
11. Lin Z, He R, Luo H, et al. RETRACTED ARTICLE: integrin-β5, a miR-185-targeted gene, promotes hepatocellular carcinoma tumorigenesis by regulating β-catenin stability. J Exp Clin Cancer Res. 2018;37:17.
12. Shi Y, Wang J, Huang G, et al. A novel epithelial-mesenchymal transition gene signature for the immune status and prognosis of hepatocellular carcinoma. Hepatol Int. 2022;16:906-17.
13. Shang L, Ye X, Zhu G, et al. Prognostic value of integrin variants and expression in post-operative patients with HBV-related hepatocellular carcinoma. Oncotarget. 2017;8:76816-31.
14. Yang X, Qin C, Zhao B, et al. Long noncoding RNA and circular RNA: two rising stars in regulating epithelial-mesenchymal transition of pancreatic cancer. Front. Oncol. 2022;12:910678.
15. Ma Q, Yang F, Xiao B, Guo X. Emerging roles of circular RNAs in tumorigenesis, progression, and treatment of gastric cancer. J Transl Med. 2024;22:207.
16. Bao Z, Yang Z, Huang Z, Zhou Y, Cui Q, Dong D. LncRNADisease 2.0: an updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019;47:D1034-7.
17. Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172:393-407.
18. Hwang SY, Danpanichkul P, Agopian V, et al. Hepatocellular carcinoma: updates on epidemiology, surveillance, diagnosis and treatment. Clin Mol Hepatol. 2025;31 Suppl:S228-54.
19. Li K, Mathew B, Saldanha E, et al. New insights into biomarkers and risk stratification to predict hepatocellular cancer. Mol Med. 2025;31:152.
20. Zhai W, Lai H, Kaya NA, et al. Dynamic phenotypic heterogeneity and the evolution of multiple RNA subtypes in hepatocellular carcinoma: the PLANET study. Natl Sci Rev. 2022;9:nwab192.
21. Zhu S, Hoshida Y. Molecular heterogeneity in hepatocellular carcinoma. Hepat. Oncol. 2018;5:HEP10.
22. Chen S, Cao Q, Wen W, Wang H. Targeted therapy for hepatocellular carcinoma: challenges and opportunities. Cancer Lett. 2019;460:1-9.
23. Likhitsup A, Razumilava N, Parikh ND. Treatment for advanced hepatocellular carcinoma: current standard and the future. Clin Liver Dis. 2019;13:13-9.
24. Liu P, Kong L, Liu Y, Li G, Xie J, Lu X. A key driver to promote HCC: cellular crosstalk in tumor microenvironment. Front. Oncol. 2023;13:1135122.
25. Farzaneh Z, Vosough M, Agarwal T, Farzaneh M. Critical signaling pathways governing hepatocellular carcinoma behavior; small molecule-based approaches. Cancer Cell Int. 2021;21:208.
26. Huang L, Lu Y, He R, et al. N4-acetylcytidine modification of ITGB5 mRNA mediated by NAT10 promotes perineural invasion in pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res. 2025;44:103.
27. Wen X, Chen S, Chen X, et al. ITGB5 promotes innate radiation resistance in pancreatic adenocarcinoma by promoting DNA damage repair and the MEK/ERK signaling pathway. Front. Oncol. 2022;12:887068.
28. Liu D, Cao J, Hu K, Peng Z. Integrin β5 interacts with G3BP1 through activating FAK/Src signaling pathway to promote gastric carcinogenesis. Sci Rep. 2025;15:28633.
29. Mccormick B, Craig HE, Chu JY, et al. A negative feedback loop regulates integrin inactivation and promotes neutrophil recruitment to inflammatory sites. J Immunol. 2019;203:1579-88.
30. Cooper J, Giancotti FG. Integrin signaling in cancer: mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell. 2019;35:347-67.
31. Juratli MA, Zhou H, Oppermann E, et al. Integrin α2 and β1 cross-communication with mTOR/AKT and the CDK-cyclin axis in hepatocellular carcinoma cells. Cancers. 2022;14:2430.
32. Jafarnejad M, Sové RJ, Danilova L, et al. Mechanistically detailed systems biology modeling of the HGF/Met pathway in hepatocellular carcinoma. npj Syst Biol Appl. 2019;5:29.
33. Iovanna J, Estaras M, Grasso D, Fernández Zapico ME, Neira JL, Santofimia-castaño P. Oncogenic stress response mechanisms as new therapeutic targets in cancer treatment: A review. Medicine. 2025;104:e42857.
34. Qiu B, Simon MC. Oncogenes strike a balance between cellular growth and homeostasis. Semin Cell Dev Biol. 2015;43:3-10.
35. Spence HJ, Dhillon AS, James M, Winder SJ. Dystroglycan, a scaffold for the ERK-MAP kinase cascade. EMBO Rep. 2004;5:484-9.
36. Quan J, Wan Z, Wu W, et al. Classical biomarkers and non-coding RNAs associated with diagnosis and treatment in gastric cancer. Oncol Res. 2025;33:1069-89.
37. Deng X, Liao T, Xie J, et al. The burgeoning importance of PIWI-interacting RNAs in cancer progression. Sci. China Life Sci. 2023;67:653-62.
38. Xu S, He L, Chen Y, et al. Clinical implications of miR-195 in cancer: mechanisms, potential applications, and therapeutic strategies. J Cancer Res Clin Oncol. 2025;151:148.
39. Chen J, Wang F, Xu H, et al. Long non-coding RNA SNHG1 regulates the Wnt/β-catenin and PI3K/AKT/mTOR signaling pathways via EZH2 to affect the proliferation, apoptosis, and autophagy of prostate cancer cell. Front. Oncol. 2020;10:552907.
40. Zeng H, Zhou S, Cai W, Kang M, Zhang P. LncRNA SNHG1: role in tumorigenesis of multiple human cancers. Cancer Cell Int. 2023;23:198.
41. Lu Y, Yu Q, Liu JH, et al. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J Biol Chem. 2003;278:40057-66.
42. Azzi A. SHIP2 inhibition alters redox‐induced PI3K/AKT and MAP kinase pathways via PTEN over‐activation in cervical cancer cells. FEBS Open Bio. 2020;10:2191-205.
43. Huang XX, Zhang Q, Hu H, et al. A novel circular RNA circFN1 enhances cisplatin resistance in gastric cancer via sponging miR‐182‐5p. J Cell Biochem. 2020;122:1009-20.
44. Sameti P, Tohidast M, Amini M, Bahojb Mahdavi SZ, Najafi S, Mokhtarzadeh A. The emerging role of MicroRNA-182 in tumorigenesis; a promising therapeutic target. Cancer Cell Int. 2023;23:134.
45. Wischmann F, Troschel FM, Frankenberg M, et al. Tumor suppressor miR-218 directly targets epidermal growth factor receptor (EGFR) expression in triple-negative breast cancer, sensitizing cells to irradiation. J Cancer Res Clin Oncol. 2023;149:8455-65.
46. Passaro A, Al Bakir M, Hamilton EG, et al. Cancer biomarkers: emerging trends and clinical implications for personalized treatment. Cell. 2024;187:1617-35.
47. Piergentili R, Gullo G, Basile G, et al. Circulating miRNAs as a tool for early diagnosis of endometrial cancer-implications for the fertility-sparing process: clinical, biological, and legal aspects. Int J Mol Sci. 2023;24:11356.
48. Samaržija I, Dekanić A, Humphries JD, et al. Integrin crosstalk contributes to the complexity of signalling and unpredictable cancer cell fates. Cancers. 2020;12:1910.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.