


Impact of metabolic reprogramming on the immune response in hepatocellular carcinoma
 0
0  ,
, Abstract
Hepatocellular carcinoma (HCC) is a highly lethal malignancy worldwide and is characterized by a low rate of early detection. In recent years, immune checkpoint inhibitors (ICIs) have been increasingly incorporated into the management of advanced HCC. However, overall response rates remain modest, indicating that immune checkpoint blockade alone is insufficient to overcome the intrinsic immunosuppressive state of HCC. Advances in metabolomics have provided new insights into early detection and therapeutic response evaluation in HCC and have underscored the functional significance of tumor metabolism in disease progression. The metabolic landscape of the HCC tumor microenvironment is predominantly shaped by alterations in glucose, lipid, and amino acid metabolism. These pathways not only support tumor cell energy production and biosynthetic demands but also reprogram local nutrient availability and metabolite composition, thereby continuously reshaping the immune milieu. This metabolic remodeling impairs effector immune cell function and facilitates the establishment and maintenance of immunosuppressive cell populations. Accordingly, this review summarizes the role of metabolic reprogramming in tumor immune regulation during HCC development, with a focus on the heterogeneity of metabolic reprogramming and immune regulation across distinct etiological backgrounds. Systematic elucidation of immunometabolic crosstalk may enhance the precision and translational potential of combination therapeutic strategies.
Keywords
INTRODUCTION
Primary liver cancer is the sixth most frequently diagnosed malignancy worldwide and the third leading cause of cancer-related mortality[1]. Hepatocellular carcinoma (HCC), the predominant histological subtype, typically has an insidious onset and progresses rapidly. As a result, nearly half of patients are no longer candidates for curative therapy at diagnosis and therefore require systemic targeted therapy[2]. However, the liver’s immunotolerant microenvironment substantially limits the therapeutic efficacy of targeted agents[3,4]. In recent years, immune checkpoint inhibitors (ICIs) have emerged as an important therapeutic option for advanced HCC[5,6]. Even so, overall response rates remain modest and are often accompanied by primary or acquired resistance, a lack of robust predictive biomarkers, and immune-related adverse events. Increasing evidence indicates that metabolic reprogramming within the tumor microenvironment (TME) plays a pivotal role in immune evasion and immunotherapy resistance in HCC[7]. HCC cells commonly exhibit aberrant activation of glucose, lipid, and amino acid metabolic pathways[8]. Glucose metabolic reprogramming, characterized by preferential glucose utilization by tumor cells and lactate accumulation, compromises the metabolic fitness of effector immune cells and acts in concert with multiple immunosuppressive signaling pathways. Dysregulated lipid metabolism further shapes a lipid-rich TME, rendering effector T (Teff) cells more susceptible to functional impairment while conferring a relative metabolic advantage on immunosuppressive cell populations. In parallel, altered amino acid metabolism reinforces immunosuppressive programs and restricts the plasticity of antitumor immune responses by modulating the availability of key nutrients and downstream signaling networks. Collectively, coordinated remodeling of glucose, lipid, and amino acid metabolism helps maintain a dynamic equilibrium within the TME. Disruption of this equilibrium provides a fundamental metabolic basis for immune evasion and immunotherapy resistance in HCC. Accordingly, this review focuses on the mechanistic roles of glucose, lipid, and amino acid metabolism in TME regulation, with the aim of elucidating the metabolic basis of HCC initiation and progression and providing a conceptual framework for optimizing immunotherapeutic strategies.
IMPACT OF GLUCOSE METABOLIC REPROGRAMMING ON THE IMMUNE RESPONSE IN HCC
Glucose is the primary energy source for cellular activities and organismal development, and glucose metabolism provides essential precursors for macromolecular biosynthesis[9]. During HCC initiation and progression, tumor cells reprogram glucose metabolism to meet their metabolic demands. In addition to supporting tumor cell growth, glucose metabolic reprogramming reshapes the immunometabolic landscape of the TME, thereby impairing antitumor immune responses [Figure 1]. Accordingly, aberrant glucose metabolism has emerged as a critical metabolic basis for immune evasion in HCC, and its role in TME regulation warrants particular attention.
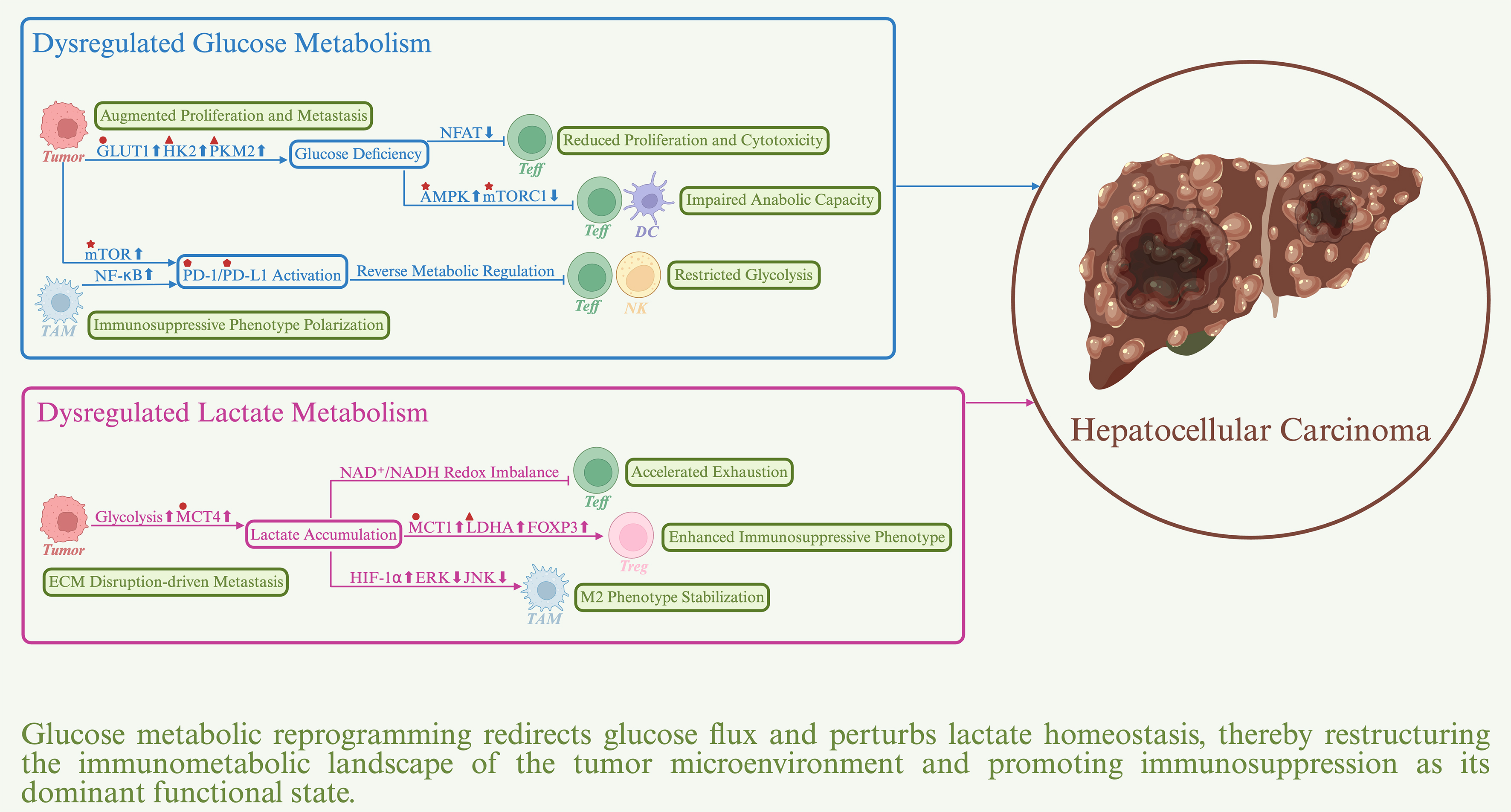
Figure 1. Impact of glucose metabolic reprogramming on the immune response in hepatocellular carcinoma. Sharp arrows denote positive regulation, whereas blunt arrows denote negative regulation. Upward symbols represent upregulation, whereas downward symbols represent downregulation. Major actionable targets are indicated by distinct geometric symbols: triangles represent metabolic enzymes, circles represent transporters, stars represent kinases, and pentagons represent immune checkpoint proteins. AMPK: AMP-activated protein kinase; GLUT1: glucose transporter 1; HK2: hexokinase 2; LDHA: lactate dehydrogenase A; MCT1: monocarboxylate transporter 1; MCT4: monocarboxylate transporter 4; mTOR: mechanistic target of rapamycin; mTORC1: mechanistic target of rapamycin complex 1; PD-1: programmed cell death protein-1; PD-L1: programmed cell death-ligand 1; PKM2: pyruvate kinase M2.
Dysregulated glucose metabolism impairs immune responses in HCC
Glucose metabolic reprogramming in HCC is characterized by the Warburg effect, in which glycolysis remains the predominant pathway supporting bioenergetic and biosynthetic demands even under aerobic conditions[10]. This metabolic shift is mediated by coordinated mechanisms, including upregulation of glucose transporter 1 (GLUT1) to increase glucose uptake, ectopic expression and activation of hexokinase 2 (HK2) in place of the physiological isoform HK4 to enhance glucose phosphorylation efficiency, and elevated expression of pyruvate kinase M2 (PKM2) to augment glycolytic flux[11,12]. These alterations are driven by aberrant activation of the Wnt/β-catenin and PI3K/AKT/mTOR signaling pathways and are further reinforced under hypoxia through hypoxia-inducible factor-1 alpha (HIF-1α)-mediated responses[13,14]. Collectively, these changes establish a metabolic program that supports rapid tumor cell proliferation and metabolic flexibility. A major immunosuppressive consequence of the Warburg effect is glucose competition, which represents a central mechanism of immune evasion in HCC. Preferential glucose uptake by tumor cells markedly limits energy availability to CD8+ T cells and suppresses their effector functions, including immunological synapse formation, target cell killing, and clonal expansion[15]. This suppression occurs because T cell activation depends on the glycolytic intermediate phosphoenolpyruvate to sustain the T cell receptor (TCR)-mediated Ca2+/nuclear factor of activated T cells (NFAT) axis[16]. Glucose deprivation disrupts NFAT nuclear translocation and subsequent transcription of effector genes. Restoring glucose metabolic balance has been shown to enhance antitumor immune responses. For example, interferon-alpha (IFN-α) suppresses HIF-1α-driven glycolysis within the TME, thereby activating the mTOR/forkhead box M1 (FOXM1) axis in CD8+ T cells and upregulating CD27 expression, ultimately sensitizing HCC to ICIs[17]. Similarly, inhibition of the deubiquitinase USP14, which stabilizes GLUT1 and promotes glucose uptake, represents a promising strategy to potentiate immunotherapy[18]. Under metabolic stress or glucose scarcity, levels of the glycolytic intermediate fructose-1,6-bisphosphate (FBP) decline markedly, leading to activation of AMP-activated protein kinase (AMPK) signaling[19,20]. AMPK activation induces dissociation of mechanistic target of rapamycin complex 1 (mTORC1) from the lysosome and subsequent inactivation, thereby impairing the anabolic metabolism required for cytotoxic CD8+ T cells and dendritic cells (DCs) to sustain immune effector functions and antigen presentation. Consequently, precise modulation of the AMPK/mTORC1 signaling pathway may represent an important strategy to restore immunometabolic balance and improve immunotherapeutic responsiveness in HCC. In addition, dysregulated glucose metabolism modulates immune checkpoint signaling and reinforces the immunosuppressive TME. Sorafenib-resistant HCC cells exhibit enhanced glycolysis through mTOR signaling, accompanied by upregulation of programmed cell death-ligand 1 (PD-L1) expression[21]. Pharmacological inhibition of mTOR activation with rapamycin restores autophagic activity and downregulates PD-L1 expression, providing a rationale for dual targeting of metabolic pathways and immune checkpoints in drug-resistant HCC. Similarly, hyaluronan fragments derived from HCC cells induce tumor-associated macrophages (TAMs) to upregulate 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), a rate-limiting glycolytic enzyme, thereby activating nuclear factor-kappa B (NF-κB) signaling and PD-L1 expression and generating a localized immune-privileged niche[22]. PD-L1 signaling further promotes the immunosuppressive polarization of TAMs and amplifies immune evasion[23]. Notably, immune checkpoint molecules can reciprocally regulate metabolic activity in immune cells. Upregulation of programmed cell death protein-1 (PD-1) and cytotoxic T lymphocyte antigen-4 (CTLA-4) cooperatively suppresses glycolysis in Teff cells, whereas PD-1 also enhances fatty acid oxidation (FAO) and lipolysis[24,25]. This metabolic reprogramming limits Teff cell function and exacerbates the effects of metabolic competition. Comparable mechanisms have been observed in tumor-infiltrating natural killer (NK) cells in HCC, indicating that coordinated dysregulation of glucose metabolism and immune checkpoint signaling sustains an immunoevasive microenvironment across multiple immune cell types[26].
Dysregulated lactate metabolism impairs immune responses in HCC
In HCC, the Warburg effect drives excessive lactate production. Lactate is actively exported from tumor cells via monocarboxylate transporter 4 (MCT4), leading to markedly elevated extracellular lactate concentrations within the TME, which often reach 30-40 mM[27,28]. Sustained lactate accumulation causes pronounced acidification of the TME and disrupts extracellular matrix architecture. These changes promote tumor cell migration and proliferation, induce angiogenesis, and enhance drug efflux and therapeutic tolerance. Importantly, lactate accumulation also interferes with antitumor immune responses through multiple mechanisms, thereby promoting HCC progression. Elevated lactate levels suppress the effector function of immune cells within the TME. High lactate concentrations markedly inhibit the proliferation, cytokine secretion, and cytotoxic activity of CD8+ T cells[29,30]. Mechanistically, lactate uptake via MCT1 induces intracellular acidification and disrupts nicotinamide adenine dinucleotide (NAD+/NADH) redox homeostasis, thereby inhibiting key glycolytic enzymes, including GAPDH and PGDH. In addition, lactate-driven histone histone H3 lysine 18 (H3K18) lactylation promotes the expression of major vault protein (MVP), which competitively inhibits β-transducin repeat-containing protein (β-TRCP)-mediated ubiquitination and degradation of PD-L1, further weakening the antitumor activity of Teff cells[31]. Accumulating evidence indicates that excessive lactate is a critical driver of T cell exhaustion in HCC[32]. Notably, lower lactate concentrations may partially restore Teff cell function, suggesting potential avenues for metabolic modulation of antitumor immunity[33]. Beyond T cells, lactate downregulates the activating receptor NKp46 on NK cells and reduces the production of IFN-γ, perforin, and granzyme B[34]. DCs are also affected, as lactate impairs their maturation and activation, thereby limiting the expression of costimulatory molecules and proinflammatory cytokines[35]. Accordingly, targeting lactate metabolism is emerging as an important strategy to alleviate effector immune cell suppression in HCC[36]. Lactate accumulation also facilitates protumor immune functions within the TME. Regulatory T (Treg) cells exhibit pronounced metabolic plasticity and preferentially adopt a lactate-fueled metabolic program by downregulating glucose uptake, upregulating MCT1-mediated lactate transport, and enhancing lactate dehydrogenase A (LDHA) activity[37]. These lactate-adapted Treg cells display enhanced immunosuppressive capacity. Moreover, lactate-induced lactylation of K72 on membrane-organizing extension spike protein (Moesin) activates downstream SMAD family member 3 (SMAD3) signaling, thereby promoting FOXP3 expression and stabilizing the immunosuppressive Treg cell phenotype[38]. In TAMs, lactate promotes polarization toward the M2 phenotype through HIF-1α signaling, accompanied by upregulation of vascular endothelial growth factor (VEGF), arginase 1 (ARG1), and interleukin-10 (IL-10)[39]. In addition, histone H3K18 lactylation enhances the expression of nuclear protein 1 (NUPR1), which sustains the M2 phenotype by suppressing ERK and JNK signaling pathways and ultimately exacerbates CD8+ T cell exhaustion[40]. Interestingly, the gut-derived metabolite D-lactate has been implicated in driving TAM repolarization toward a proinflammatory M1 phenotype[41]. However, whether D-lactate contributes to hepatic immune modulation via the portal circulation remains to be elucidated.
Etiology-associated heterogeneity of glucose metabolic reprogramming and immune regulation in HCC
HCC shows etiology-associated heterogeneity in glucose metabolic reprogramming. In virus-related HCC, viral proteins directly enhance key glycolytic nodes, thereby promoting a shift toward a Warburg phenotype. Both hepatitis B virus (HBV)- and HCV-related HCC exhibit markedly increased HK2 activity. In HBV-related HCC, this increase is driven by hepatitis B virus X protein (HBx)-mediated activation of the NF-κB p65/HK2 axis. In HCV-related HCC, it arises from a direct interaction between HCV nonstructural protein 5A (NS5A) and HK2[42,43]. In addition, HBV pre-S2 deletion mutants activate the mTOR signaling cascade, thereby upregulating GLUT1 expression[44]. These mechanisms may help explain the convergence between HBV- and HCV-related HCC in the transcriptomic landscape and immune profiles[45]. Notably, the higher prevalence of β-catenin mutations in HCV-related HCC predisposes tumor cells to a hepatocyte-like subtype characterized by reduced glycolysis and enhanced FAO[46,47]. This metabolic shift is typically accompanied by downregulation of chemokine expression, impaired DC recruitment, and reduced CD8+ T cell infiltration, resulting in an immune-excluded TME[48]. By contrast, HBV-related HCC more commonly shows preserved immune cell infiltration with functional suppression, whereas HCV-related HCC exhibits more pronounced metabolic and immunological stratification[49]. In alcohol-related HCC, the association between β-catenin mutation and an immune-excluded phenotype appears more prominent, while the immunosuppressive effects mediated by dysregulated glucose metabolism are comparatively attenuated[50]. In metabolic dysfunction-associated steatotic liver disease (MASLD)/metabolic dysfunction-associated steatohepatitis (MASH)-related HCC, metabolic reprogramming preferentially involves aberrant activation of lactate metabolism. Elevated expression of GLUT1 and MCT4 correlates with increased tumor invasiveness and adverse clinical outcomes, although the upstream regulatory mechanisms remain incompletely defined[51]. Compared with virus-related HCC, MASLD/MASH-related HCC may be enriched for specific nonviral driver genetic events. For example, activin A receptor type 2A (ACVR2A) loss occurs more frequently in MASLD/MASH-related HCC and enhances lactate production and efflux through suppression of SMAD signaling, followed by upregulation of LDHA and MCT4[52]. Furthermore, MASH-related HCC more readily establishes a specialized spatial interaction network among TAMs, MDSCs, and CD8+ T cells, suggesting that myeloid cell-mediated TME remodeling constitutes a principal mechanism of immune suppression[53]. Given the limited benefit of ICIs in MASH-related HCC, therapeutic strategies aimed at dismantling myeloid cell-driven spatial immune sequestration may be critical for enhancing immunotherapeutic responsiveness[54].
IMPACT OF LIPID METABOLIC REPROGRAMMING ON THE IMMUNE RESPONSE IN HCC
Lipids serve not only as fundamental energy sources and structural components of cellular membranes but also as key signaling mediators that regulate cell growth, apoptosis, and immune responses[55]. In HCC, lipid metabolic dysregulation associated with metabolic reprogramming profoundly reshapes energy allocation and immune architecture within the TME, thereby promoting immune evasion [Figure 2].
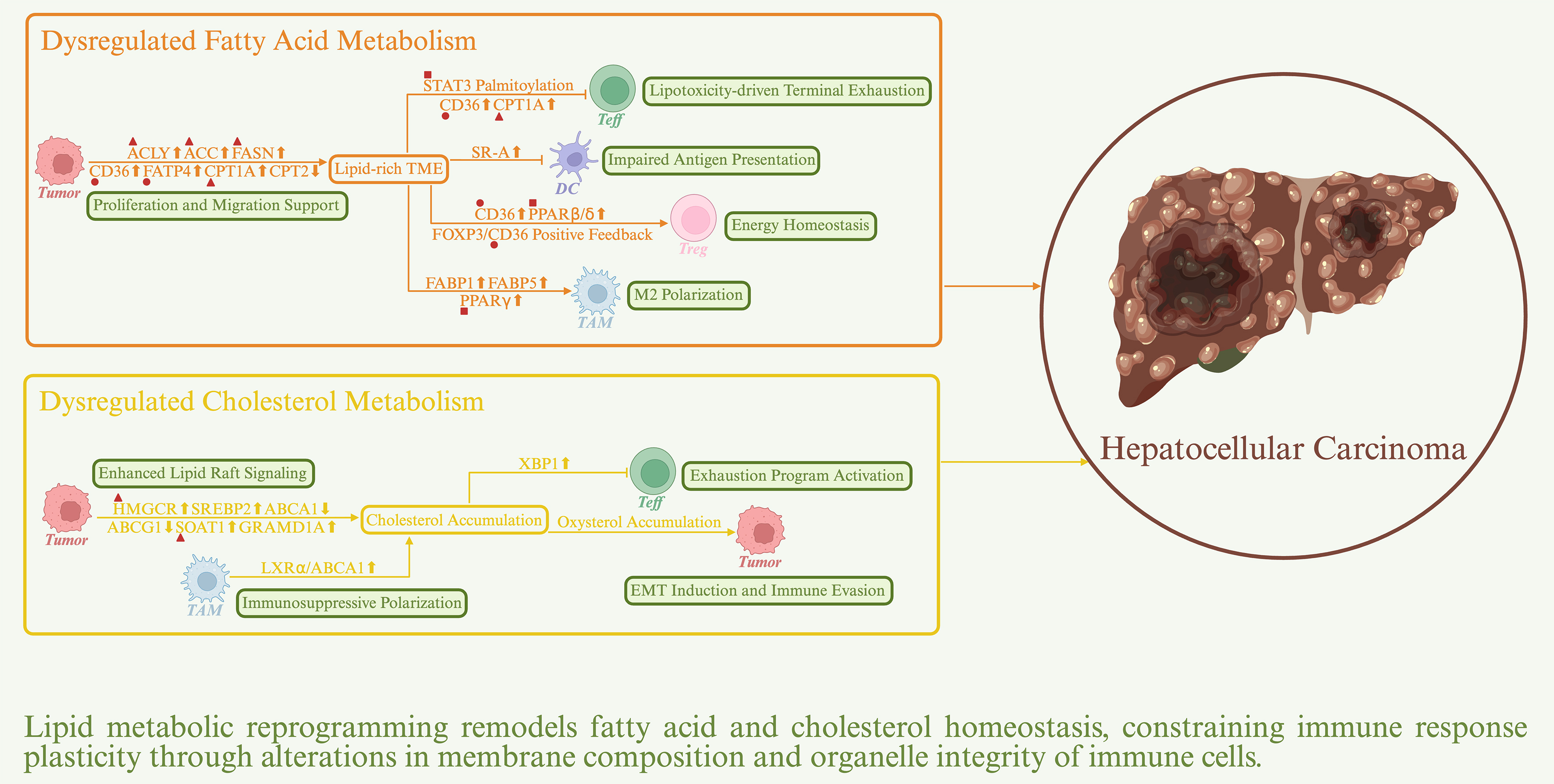
Figure 2. Impact of lipid metabolic reprogramming on the immune response in HCC. Sharp arrows denote positive regulation, whereas blunt arrows denote negative regulation. Upward symbols represent upregulation, whereas downward symbols represent downregulation. Major actionable targets are indicated by distinct geometric symbols: triangles represent metabolic enzymes, circles represent transporters, and squares represent transcription factors. ACC: Acetyl-CoA carboxylase; ACLY: ATP citrate lyase; CD36: cluster of differentiation 36; CPT1A: carnitine palmitoyltransferase 1A; FASN: fatty acid synthase; FATP4: fatty acid transport protein 4; HMGCR: 3-hydroxy-3-methylglutaryl-CoA reductase; PPARβ/δ: peroxisome proliferator-activated receptor β/δ; PPARγ: peroxisome proliferator-activated receptor γ; SOAT1: sterol O-acyltransferase 1; STAT3: signal transducer and activator of transcription 3.
Dysregulated fatty acid metabolism impairs immune responses in HCC
HCC cells undergo progressive metabolic reprogramming to establish a dynamically coordinated network of fatty acid synthesis (FAS), uptake, and oxidation[56]. This network is tightly regulated to meet the demands imposed by a hypoxic and nutrient-deprived TME. Enhanced de novo lipogenesis, driven by upregulation of ATP citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FASN), is regulated by the PI3K/AKT/mTORC1 signaling pathway through activation of sterol regulatory element-binding protein 1 (SREBP1)[57,58]. This regulatory circuitry enables tumor cells to preserve membrane integrity and sustain prosurvival signaling under metabolic stress. Concurrently, elevated expression of CD36 and fatty acid transport protein 4 (FATP4) enhances exogenous lipid uptake, thereby alleviating biosynthetic pressure while modulating membrane fluidity to promote tumor cell motility and invasiveness[59]. Carnitine palmitoyltransferase 1A (CPT1A) and CPT2 regulate energy production and lipid load balance by controlling FAO, a process critical for HCC proliferation, redox homeostasis, and metabolic adaptation[60,61]. As a consequence of increased lipid synthesis and catabolism in HCC cells and associated stromal compartments, together with chronic inflammation and hypoxia-driven lipid mobilization, the HCC TME becomes enriched in fatty acids[62]. Within this lipid-rich TME, CD8+ T cells lack sufficient enzymatic capacity to efficiently catabolize excess fatty acids, leading to lipotoxic accumulation and subsequent functional exhaustion[63]. Notably, epigenetic silencing of the E3 ubiquitin ligase Riplet in HCC aberrantly enhances FAS in tumor cells, resulting in excessive palmitate production that drives terminal CD8+ T cell exhaustion through signal transducer and activator of transcription 3 (STAT3) palmitoylation-dependent signaling[64]. Prolonged energy deprivation further induces metabolic reprogramming in CD8+ T cells, characterized by increased CD36 expression and enhanced uptake of oxidized low-density lipoprotein (oxLDL). This process activates p38/CCAAT enhancer-binding protein β (CEBPβ) signaling, leading to upregulation of transferrin receptor 1 (TFR1), increased intracellular iron accumulation, and heightened oxidative stress. These changes culminate in lipid peroxidation and accelerated T cell dysfunction[65]. In parallel, the PD-1/STAT3 signaling pathway promotes FAO by upregulating CPT1A while suppressing glycolysis, thereby further weakening the antitumor activity of CD8+ T cells[66]. Collectively, STAT3 represents a critical signaling node linking fatty acid metabolism to immunosuppression and constitutes a potential therapeutic target in HCC[67]. Compared with Teff cells, Treg cells exhibit greater metabolic plasticity and adaptability. In lipid-rich environments, tumor-infiltrating Treg cells markedly upregulate CD36, thereby enhancing fatty acid uptake and activating peroxisome proliferator-activated receptor β/δ (PPARβ/δ) signaling, which drives FAO and mitochondrial biogenesis[68]. This metabolic reprogramming enables Treg cells to maintain energy homeostasis and immunosuppressive capacity within the lactate-rich and glucose-deprived microenvironment. Moreover, FOXP3 directly activates CD36 transcription, reinforcing this lipid-driven immunoregulatory phenotype and further impairing antitumor immunity[69]. Accumulating evidence supports CD36 as a promising therapeutic target for enhancing immunotherapeutic responsiveness in HCC[70]. Fatty acid metabolic remodeling in myeloid cells further contributes to immune evasion. In DCs, a lipid-rich environment promotes lipotoxic accumulation and markedly impairs antigen processing and cross-presentation[71]. Upregulation of scavenger receptor-A (SR-A) facilitates excessive lipid uptake and further dampens DC-mediated activation of adaptive immunity[72]. In TAMs, fatty acid-binding protein 1 (FABP1) is markedly upregulated during HCC progression and cooperates with PPARγ to activate CD36-dependent FAO, thereby metabolically supporting M2 polarization[73]. In late-stage tumors characterized by hypoxia and necrosis, HCC-derived exosomes deliver FABP5 to TAMs, thereby activating PPARγ and suppressing PPARα-mediated FAO. This metabolic shift favors lipid storage and lipid droplet accumulation, stabilizing the immunosuppressive M2 phenotype[74]. These findings indicate that FABP-mediated lipid metabolic plasticity dynamically regulates TAM polarization and immune evasion through spatiotemporally controlled mechanisms. In MDSCs, FAO flux is likewise markedly elevated, whereas genetic deletion of CD36 or pharmacological inhibition of STAT3 significantly attenuates their immunosuppressive activity[75]. Of particular clinical relevance, sorafenib, a frontline therapeutic for HCC, exhibits a nonclassical resistance mechanism by activating PPARα signaling and enhancing FAO flux in MDSCs[76]. This metabolic reprogramming not only drives the expression of immunosuppressive mediators but also impairs MDSC differentiation into macrophages, ultimately dampening antitumor immunity.
Dysregulated cholesterol metabolism impairs immune responses in HCC
Aberrant cholesterol metabolism, particularly overactivation of de novo cholesterol biosynthesis, is a hallmark of HCC and contributes to enhanced lipid raft-associated signaling in tumor cell membranes[77,78]. Elevated expression of 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) promotes tumor proliferation by sustaining c-Myc and FOXM1 signaling, whereas persistent activation of SREBP2 facilitates epithelial-mesenchymal transition (EMT) and promotes immune evasion. Sustained activation of the cholesterol biosynthetic pathway concurrently suppresses cholesterol efflux, as reflected by impaired transcriptional activity of liver X receptors (LXRs) and marked downregulation of the downstream transporters ABCA1 and ABCG1[79,80]. This suppression disrupts intracellular cholesterol distribution and metabolic feedback regulation, establishing a self-reinforcing positive feedback loop that further amplifies cholesterol biosynthesis. In addition, cholesterol esterification mediated by sterol O-acyltransferase 1 (SOAT1) and intracellular redistribution driven by GRAM domain-containing protein 1A (GRAMD1A) exacerbate cholesterol retention and recirculation, ultimately promoting the formation of a cholesterol-rich TME[81,82]. Excessive cholesterol accumulation induces CD8+ T cell exhaustion. Mechanistically, cholesterol overload disrupts lipid metabolic homeostasis in CD8+ T cells, triggers endoplasmic reticulum (ER) stress, and activates the ER stress sensor XBP1[83]. XBP1 subsequently drives the expression of immunosuppressive receptors, including PD-1, TIM-3, and LAG-3, thereby impairing antitumor immunity. Conversely, pharmacological inhibition of SOAT1 to block cholesterol esterification increases free cholesterol in the plasma membrane, alleviates ER stress, enhances TCR signaling, and promotes immunological synapse formation, thereby markedly augmenting CD8+ T cell cytotoxicity[84]. Unlike fatty acids, cholesterol often acts as a proinflammatory mediator in TAMs. Despite elevated systemic cholesterol levels in the HCC TME, intracellular cholesterol content in TAMs is markedly reduced[85,86]. This paradox largely reflects tumor-derived immunosuppressive signals that activate the LXRα/ABCA1 axis, thereby enhancing cholesterol efflux and disrupting intracellular cholesterol homeostasis. This metabolic reprogramming maintains TAMs in a low-cholesterol, semimature state characterized by impaired antigen-presenting capacity and a predominantly immunosuppressive phenotype. Notably, exogenous cholesterol can synergize with tumor-derived signals to further reinforce this immunosuppressive reprogramming. In MDSCs, although mechanistic studies remain limited, available evidence indicates that cholesterol homeostasis is a key determinant of immunosuppressive activity. Genetic ablation of cholesterol efflux-related genes attenuates immunosuppression[87]. Moreover, activation of LXRβ by LXR agonists upregulates apolipoprotein E (ApoE) and cholesterol efflux pathways, leading to dysregulated lipid efflux and induction of ApoE/low-density lipoprotein receptor-related protein 8 (LRP8)-dependent apoptosis in MDSCs, thereby restoring antitumor immunity[88,89]. Future studies should delineate the dynamic regulation of cholesterol homeostasis to enable precise modulation of the immunometabolic network in HCC. High-cholesterol conditions also promote pathological accumulation of oxysterols, which further reinforces the immunosuppressive milieu in HCC. Mechanistic studies indicate that accumulation of 25-hydroxycholesterol (25-HC) activates NF-κB and STAT3 signaling, increases FABP4 expression, and promotes matrix metalloproteinase (MMP) release, thereby enhancing tumor cell migration and invasiveness[90]. In addition, 27-HC activates LXR signaling to modulate inflammatory cytokine profiles in TAMs and lymphocytes, while concurrently triggering the NF-κB/Twist1 axis to induce EMT and potentiate immune evasion[91].
Etiology-associated heterogeneity of lipid metabolic reprogramming and immune regulation in HCC
Distinct etiological contexts engage different upstream regulatory networks, thereby shaping discrete lipid metabolic subtypes. MASLD/MASH is frequently accompanied by pathological lipid accumulation, which impairs mitochondrial function and activates cell death signaling pathways[92]. To adapt to lipotoxic stress, HCC cells downregulate CPT2 to suppress FAO, thereby attenuating Src/JNK-mediated apoptotic signaling and gaining a survival advantage[93]. Concurrently, oxLDL uptake induces lipophagy, reprograms intracellular lipid flux, and activates a yes-associated protein (YAP)-dependent proproliferative transcriptional program[94]. Upregulation of SOAT1 enhances cholesterol esterification and lipid droplet storage and, through SREBP2-mediated feedback regulation, remodels membrane cholesterol distribution, ultimately promoting EMT and metastasis[95]. Collectively, in MASLD/MASH-related HCC, lipid metabolic reprogramming may establish a positive feedback loop that shifts from metabolic adaptation to metabolic dependency, thereby facilitating lipid accumulation-associated immune evasion. Lipid-rich microenvironments induce chronic inflammation and drive aberrant immune activation, which exacerbates hepatic fibrosis and tissue injury and ultimately promotes tumorigenesis[96]. MASLD/MASH-related HCC exhibits heterogeneous immune regulatory patterns, giving rise to subpopulations with differential immunotherapeutic responses. For example, an endothelial cell-dominant subset characterized by high FABP4 expression promotes vascular normalization and enhances CD8+ T cell infiltration[97]. In contrast, an intratumoral steatotic phenotype displays an immune-enriched yet functionally suppressed TME marked by T cell exhaustion and enhanced immune checkpoint signaling[98]. These observations provide a basis for precision stratification. In contrast to MASLD/MASH-related HCC, lipid metabolic reprogramming in HBV-related HCC is directly driven by viral factors. HBx modulates SREBP-mediated lipogenic pathways while enhancing autophagic flux, thereby coordinately supporting viral persistence and tumor progression[99,100]. Moreover, a high-risk subtype of early-stage HBV-related HCC characterized by elevated SOAT1 expression and dysregulated cholesterol metabolism has been associated with poor prognosis[82]. In HCV-related HCC, lipid metabolic remodeling preferentially involves diacylglycerol O-acyltransferase 1 (DGAT1)-mediated lipid droplet biogenesis, enhanced viral protein localization, and suppressed triglyceride turnover, thereby establishing a lipid droplet-dependent metabolic niche that sustains viral replication and tumor progression[101]. Although immune cells may initially exhibit heightened reactivity in the context of viral infection, lipid metabolic dysregulation frequently drives their transition toward an exhausted state[102]. In alcohol-related HCC, ethanol oxidation leads to abnormal NADH accumulation, thereby promoting lipogenesis and suppressing FAO[103]. Lipid peroxidation products further impair mitochondrial function and establish a self-amplifying cycle of oxidative stress and metabolic disruption. The immune landscape of alcohol-related HCC is predominantly characterized by chronic inflammation and a myeloid-skewed protumor immune milieu[104].
IMPACT OF AMINO ACID METABOLIC REPROGRAMMING ON THE IMMUNE RESPONSE IN HCC
Amino acids are essential nutritional substrates for cellular growth and homeostasis and play critical roles in protein synthesis, energy metabolism, and signal transduction[105]. In HCC, the metabolic landscapes of glutamine, tryptophan, and arginine undergo profound reprogramming. Dysregulated amino acid metabolism actively remodels the TME, thereby promoting the establishment and amplification of an immunosuppressive milieu [Figure 3].
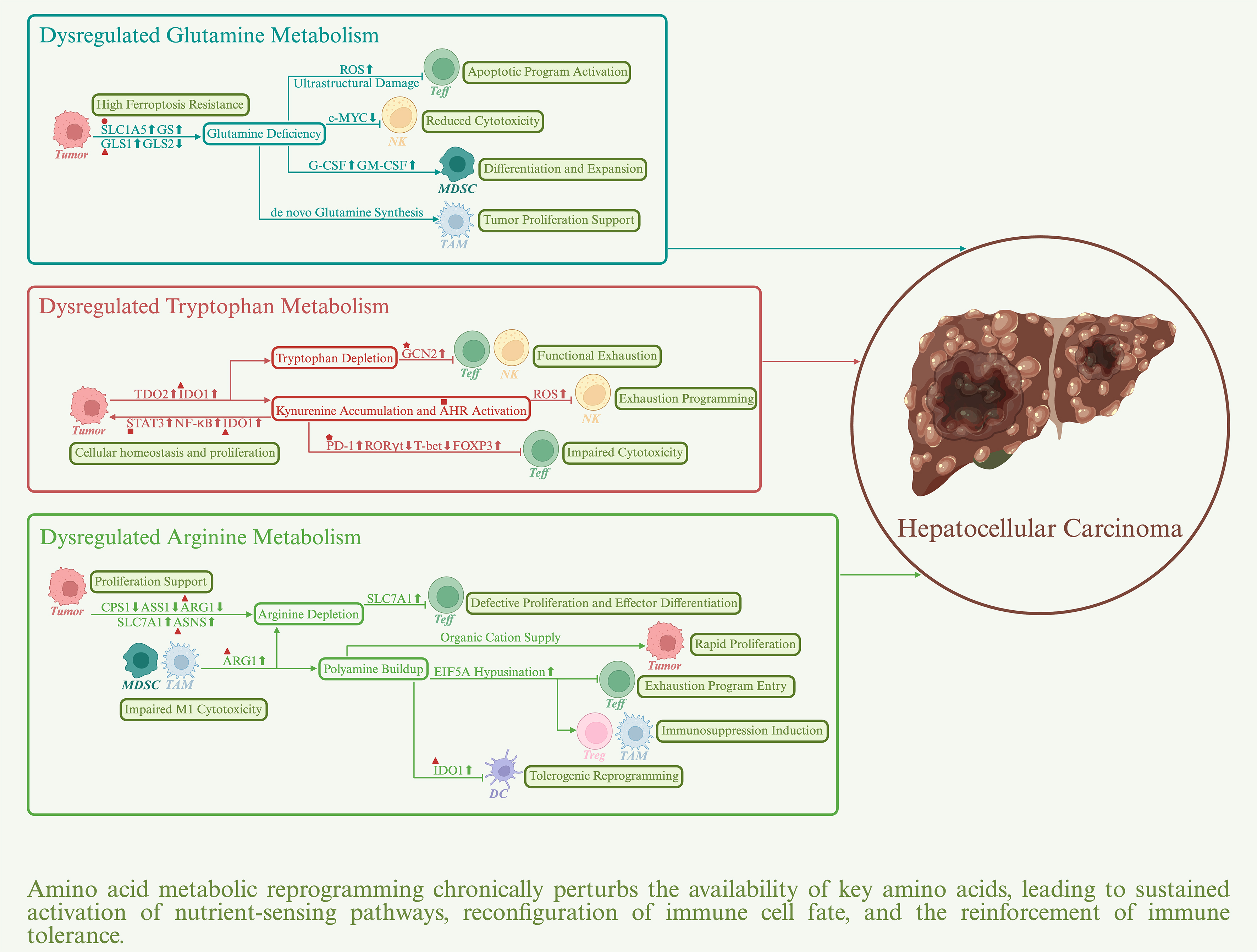
Figure 3. Impact of amino acid metabolic reprogramming on the immune response in HCC. Sharp arrows denote positive regulation, whereas blunt arrows denote negative regulation. Upward symbols represent upregulation, whereas downward symbols represent downregulation. Major actionable targets are indicated by distinct geometric symbols: triangles represent metabolic enzymes, circles represent transporters, stars represent kinases, squares represent transcription factors, and pentagons represent immune checkpoint proteins. AHR: Aryl hydrocarbon receptor; ARG1: arginase 1; ASNS: asparagine synthetase; GCN2: general control nonderepressible 2; GLS1: glutaminase 1; IDO1: indoleamine-2,3-dioxygenase 1; PD-1: programmed cell death protein-1; SLC1A5: solute carrier family 1 member 5; STAT3: signal transducer and activator of transcription 3.
Dysregulated glutamine metabolism impairs immune responses in HCC
Aberrant activation of glutamine metabolism is a hallmark of HCC[106,107]. Key components of this pathway, including the glutamine transporter SLC1A5, kidney-type glutaminase GLS1, and glutamine synthetase (GS), are frequently upregulated in HCC tissues. By contrast, GLS2, the liver-type glutaminase that maintains metabolic homeostasis in normal hepatic tissue, is often silenced or markedly downregulated. Notably, in β-catenin-mutant HCC, GS overexpression paradoxically restrains tumor progression by limiting aberrant accumulation of free glutamate and suppressing hyperactivation of mTORC1 signaling[108]. This observation underscores the importance of nitrogen flux homeostasis, governed by the balance between glutamine synthesis and catabolism, in shaping the metabolic adaptability and invasive potential of HCC cells. Beyond supporting anabolic demands, glutamine metabolism sustains HCC cell survival under persistent oxidative stress by fueling glutathione synthesis and replenishing the tricarboxylic acid (TCA) cycle[109]. These processes maintain nicotinamide adenine dinucleotide phosphate (NADPH)-dependent redox homeostasis required for lipid and protein stability, thereby conferring resistance to ferroptosis. More importantly, glutamine metabolic reprogramming contributes directly to immune evasion. Nutrient competition suppresses effector immune cell function because CD8+ T cells depend heavily on glutamine metabolism for activation. Glutamine deprivation disrupts mitochondrial homeostasis, as reflected by reduced membrane potential, accumulation of reactive oxygen species (ROS), and ultrastructural damage[110]. These alterations activate apoptotic programs, leading to downregulation of effector molecules, upregulation of inhibitory receptors, and eventual T cell exhaustion. Interestingly, systemic blockade of glutamine metabolism simultaneously impairs glycolysis and oxidative phosphorylation (OXPHOS) in tumor cells[111]. In contrast, CD8+ T cells maintain energy and redox homeostasis by upregulating acetate utilization and oxidative metabolism and are reprogrammed into a memory-like state with long-term survival potential. This divergent metabolic response suggests a strategy for selectively alleviating tumor-associated immunosuppression. Similarly, NK cells are functionally compromised under glutamine-limiting conditions because of suppressed expression of the metabolic regulator c-Myc[112]. Metabolic stress can also enhance the function of immunosuppressive cell populations. In TAMs, glutamine metabolism promotes an immunosuppressive M2 phenotype through generation of α-ketoglutarate (α-KG), which drives epigenetic remodeling. In addition, glutamate provision fuels the TCA cycle and sustains OXPHOS[113,114]. Unlike effector immune cells, TAMs retain the capacity for de novo glutamine synthesis, conferring a metabolic advantage within the nutrient-deprived TME. Persistent GS overexpression in TAMs is therefore considered essential for maintaining the M2 phenotype. Moreover, glutamine synthesized by TAMs can be utilized by HCC cells as a carbon source to support proliferation and metabolic demands. Conversely, intermediates derived from tumor glutamine metabolism can reprogram macrophages to further reinforce immunosuppressive functions. Notably, tumor-derived glutamate induces chemokine CCL2 expression, thereby promoting recruitment of C-C motif chemokine receptor 2 (CCR2+) TAMs and exacerbating HCC progression[115]. In MDSCs, severe glutamine scarcity acts as a metabolic stress signal that drives tumor cells to upregulate granulocyte colony-stimulating factor (G-CSF) and granulocyte macrophage colony-stimulating factor (GM-CSF), thereby systemically reconfiguring the TME to favor MDSC expansion and differentiation[116]. However, MDSC dependence on glutamine is highly dose- and time-dependent[117]. Systematic delineation of the underlying regulatory network in the multidimensional context of the TME remains necessary.
Dysregulated tryptophan metabolism impairs immune responses in HCC
Under physiological conditions, hepatic tryptophan catabolism proceeds primarily through the kynurenine pathway, which is catalyzed by tryptophan-2,3-dioxygenase (TDO2) to maintain systemic immunometabolic homeostasis[118]. In HCC, however, this pathway is aberrantly upregulated. Both TDO2 and indoleamine-2,3-dioxygenase 1 (IDO1) are markedly overexpressed in tumor tissues compared with normal liver. Their upregulation is driven, respectively, by oncogenic signaling pathways such as Wnt/β-catenin and NF-κB and by mediators associated with chronic inflammation. Increased expression of these enzymes correlates closely with higher tumor burden and poorer histological differentiation[119,120]. Notably, some studies have interpreted IDO1 overexpression as a metabolically induced negative feedback mechanism secondary to immune activation, rather than solely as a protumorigenic factor[121]. For example, in inflamed microenvironments enriched in CD8+ T cells, IFN-γ induces IDO1 expression in tumor cells through the JAK2/STAT1 signaling pathway, thereby restraining excessive cytotoxic responses. With tumor progression, the TME shifts toward an immune-excluded phenotype, during which IDO1 expression transitions to a sustained, tumor-intrinsic immunoevasive program that further reinforces the immunosuppressive network[122]. Persistent tryptophan depletion leads to the accumulation of uncharged transfer RNAs (tRNAs), which activates general control nonderepressible 2 (GCN2) and initiates an amino acid starvation response[123]. This response induces cell-cycle arrest in CD8+ T cells and NK cells, promotes differentiation of naïve CD4+ T cells into Treg cells, and facilitates tumor immune evasion. In parallel, kynurenine, an endogenous ligand of the aryl hydrocarbon receptor (AHR), sustains AHR activation and mediates broad immunosuppressive effects. In HCC cells, excessive AHR signaling promotes tumor cell proliferation and acquired drug resistance through STAT3 and NF-κB signaling[124,125]. This process further increases IDO1 expression and establishes a self-reinforcing positive feedback loop. Within adaptive immunity, AHR activation upregulates PD-1 and reprograms transcriptional networks associated with T cell exhaustion, thereby impairing CD8+ T cell cytotoxicity[126,127]. At the same time, AHR suppresses the transcription factors RORγt and T-bet, inhibiting T helper 1 (Th1) and Th17 differentiation while promoting FOXP3 and IL-10 expression, which collectively biases CD4+ T cells toward a Treg cell phenotype. Within innate immunity, AHR signaling downregulates key NK cell-activating receptors, including NKG2D and NKp46, and activates ROS-dependent mitochondrial apoptotic pathways, thereby promoting NK cell exhaustion[128,129]. In DCs, AHR activation reduces the expression of costimulatory molecules and IL-12 while increasing IL-10, IDO1, and inhibitory receptors such as ILT3 and ILT5, thereby promoting a tolerogenic phenotype[130]. Importantly, the immunological consequences of AHR activation are highly ligand dependent, with distinct endogenous and exogenous ligands eliciting divergent or even opposing effects[131,132]. Accordingly, comprehensive elucidation of ligand-specific AHR engagement and potential allosteric regulatory mechanisms is essential for the rational design of selective AHR modulators aimed at precise therapeutic targeting of this pathway.
Dysregulated arginine metabolism impairs immune responses in HCC
In HCC, arginine metabolism is extensively reprogrammed at the systemic level. Suppression of the urea cycle, marked by downregulation of carbamoyl phosphate synthetase 1 (CPS1) and argininosuccinate synthase 1 (ASS1), compromises endogenous arginine regeneration[133,134]. As a compensatory response, upregulation of the arginine transporter SLC7A1 enhances extracellular arginine uptake, whereas increased expression of asparagine synthetase (ASNS) facilitates arginine influx through amino acid exchange mechanisms[135]. Concurrently, reduced ARG1 expression limits arginine flux into the ornithine pathway, leading to substantial intracellular arginine accumulation[136]. Excess arginine can directly bind to and modulate the transcriptional and splicing activity of RNA-binding motif protein 39 (RBM39), thereby reshaping metabolic circuits and establishing a proproliferative positive feedback loop that sustains tumor growth[137]. Arginine depletion driven by metabolic dysregulation also has profound immunological consequences. It induces compensatory upregulation of SLC7A1 in T cells[138]. However, because T cells depend strongly on extracellular arginine and have limited capacity for de novo arginine synthesis, they exhibit impaired survival, proliferation, and effector differentiation under arginine-limiting conditions. Moreover, activated hepatic stellate cells induce ARG1 expression in C-X3-C motif chemokine receptor 1 (CX3CR1+) TAMs through retinoic acid signaling[139,140]. In parallel, CD14+ human leukocyte antigen-DR isotype (HLA-DR-/low) MDSCs, which are enriched in the HCC microenvironment, display markedly elevated arginase activity. Together, these processes further exacerbate local arginine scarcity and drive T cell hyporesponsiveness or functional exhaustion. Notably, ARG1 also competes with inducible nitric oxide synthase (iNOS) for arginine as a substrate, thereby limiting NO production and impairing the cytotoxicity of M1-like macrophages[141]. Metabolic reprogramming of arginine utilization in TAMs and MDSCs also increases polyamine synthesis, which contributes to the establishment of an immunosuppressive milieu. Polyamines enrich intracellular and extracellular compartments with organic cations that stabilize nucleic acids and ribosomes, thereby supporting the high metabolic throughput required for DNA replication, RNA transcription, and protein synthesis in proliferating HCC cells[142]. More importantly, polyamines promote hypusination of eukaryotic translation initiation factor 5A (EIF5A) and selectively enhance translation of proteins involved in OXPHOS and mitophagy[143]. This translational bias shifts immune cell metabolism toward an OXPHOS-dominant state, driving Teff cell exhaustion while promoting differentiation of Treg cells and M2-like macrophages. In addition, polyamines activate IDO1 signaling, reprogramming DCs toward a tolerogenic phenotype and reinforcing the immunosuppressive programs of MDSCs[144,145]. Accordingly, targeting dysregulated arginine metabolism in HCC is emerging as a promising therapeutic strategy. Three principal approaches have been proposed, including arginine deprivation to exploit tumor auxotrophy, augmentation of arginine bioavailability to restore immune competence, and inhibition of polyamine biosynthesis or export to counteract immunosuppression[146].
Etiology-associated heterogeneity of amino acid metabolic reprogramming and immune regulation in HCC
HBV- and HCV-related HCC exhibit marked divergence in glutamine metabolic reprogramming. HBV-related HCC predominantly depends on exogenous glutamine uptake. SLC1A5 has been identified as an independent risk factor, and its elevated expression correlates closely with enrichment of protumorigenic immune subsets and upregulation of immune checkpoint molecules[147]. By contrast, HCV-related HCC preferentially relies on endogenous glutamine synthesis, reflected by high GS expression that is more frequently observed in well-differentiated tumors[148]. GS overexpression is strongly associated with β-catenin mutations and, in the context of urea cycle impairment, constrains tumor growth signaling by maintaining nitrogen homeostasis[149]. These findings suggest that distinct glutamine metabolic phenotypes in HCV-related HCC are determined by specific molecular backgrounds. Activation of IFN-I signaling represents a key molecular feature of virus-related HCC and may promote immune evasion through amino acid metabolic reprogramming. IFN-I selectively induces hepatocellular TDO2 expression, thereby integrating tryptophan catabolism into the antiviral response program[150]. In parallel, IFN-I suppresses hepatic urea cycle flux, impairs arginine regeneration, and causes systemic hypoargininemia[151]. Notably, HCV-related HCC shows significant enrichment of IFN-stimulated genes and is more likely than HBV-related HCC to exist in a state of chronic antiviral signaling activation[152]. In MASLD/MASH-related HCC, glutamine metabolism is dynamically reprogrammed during disease progression. During hepatic fibrogenesis, stromal cell-mediated glutaminolysis via GLS1 is markedly enhanced, providing energy and biosynthetic precursors to support cellular proliferation and collagen synthesis[153]. Meanwhile, emerging tumor nodules activate GS-dependent glutamine synthesis to maintain metabolic homeostasis and sustain growth potential[154]. Cell type-specific glutamine metabolic reprogramming may facilitate metabolic coupling among stromal cells, myeloid cells, and tumor cells, thereby intensifying nutrient competition within the TME. In this context,α-KG-dependent epigenetic remodeling and OXPHOS-supported metabolic activity jointly promote M2 polarization of TAMs, conferring tolerogenic antigen-presenting functions within the TME[155]. Furthermore, both MASLD/MASH-related and alcohol-related HCC display immunosuppressive phenotypes associated with arginine metabolic dysregulation[156,157]. In MASLD/MASH-related HCC, urea cycle dysfunction secondary to metabolic disequilibrium constitutes the principal driver, whereas in alcohol-related HCC, marked expansion of ARG1+ granulocytic myeloid-derived suppressor cells (G-MDSCs) predominates. Although arginine metabolic reprogramming converges on similar terminal immunosuppressive phenotypes across etiologies, the upstream regulatory mechanisms remain heterogeneous. This mechanistic divergence provides a rationale for differential immunotherapeutic responses across etiologies and underscores the necessity of etiology-informed stratified targeting of arginine metabolic pathways.
CURRENT LIMITATIONS AND FUTURE PERSPECTIVES
A major unresolved gap is the limited ability to assign metabolism-associated signals within the TME to specific cell types and tissue niches in a causal, spatially resolved manner. Most existing studies remain restricted to correlative associations between bulk metabolite abundance and immunosuppressive phenotypes, and they lack direct evidence that defined cell populations generate or consume key metabolites within specific niches to suppress neighboring immune cells at relevant spatial scales. Recently, a metabolically interactive niche was identified at the invasive tumor front, in which cancer-associated fibroblast (CAF)-derived lactate drives macrophage polarization toward an immunosuppressive state[158]. Conventional metabolic profiling based on bulk tissue or pooled cell populations is inherently limited in capturing such spatially restricted signals. Integration of multimodal spatial omics with single-cell mass spectrometry imaging offers a promising approach to address this limitation[159,160]. Beyond spatial heterogeneity, metabolic regulation of antitumor immunity also depends on temporal dynamics and metabolic flux. Frameworks that rely on static metabolite concentrations are therefore insufficient to establish causal links between metabolism and immune regulation. During cellular functional reprogramming, changes in pathway activity often precede detectable shifts in steady-state metabolite levels, which may remain relatively stable within specific temporal windows[161]. These temporally ordered and sustained changes in metabolic activity directly govern cell state transitions but are largely overlooked by conventional static metabolomics. Incorporating stable isotope tracing into high-throughput single-cell mass spectrometry, enabling construction of a dynamic single-cell metabolomics framework, provides an experimentally tractable strategy to address this limitation[162]. Current evidence supporting metabolic control of tumor immunity is derived predominantly from in vitro models or analyses of dissociated tissues, which preclude functional validation within intact in situ microenvironments that preserve tissue architecture and cell-cell interactions. This limitation constrains reliable evaluation of non-cell-autonomous effects, physiological relevance, and translational potential. Recent advances indicate that integrating in situ clustered regularly interspaced short palindromic repeats (CRISPR)-based genetic perturbation with spatial transcriptomics on the same tissue section enables direct interrogation of how perturbation of specific metabolic pathways reshapes tumor cell states and the composition and function of surrounding immune cells while preserving spatial organization and immune niches[163]. Methodologically, this framework provides a powerful approach for in situ causal dissection linking genetic perturbations to TME remodeling.
As metabolic-immune combination strategies move toward clinical translation, substantial challenges remain. Arginine deprivation mediated by pegylated arginine deiminase (ADI-PEG 20) has demonstrated a survival benefit in the second-line treatment of advanced HCC only in a small subset of patients capable of sustaining prolonged metabolic depletion[164]. In the setting of chronic liver disease, treatment-related immunogenicity is often amplified, with anti-drug antibodies emerging early during therapy and leading to rapid attenuation of arginine depletion. In addition, pharmacokinetic variability and restricted intratumoral drug distribution jointly undermine the durability of metabolic interventions. These observations indicate that future clinical trial designs should prioritize the depth and duration of metabolic target engagement as core pharmacodynamic endpoints to determine whether an intervention persistently reaches its intended metabolic target. Similarly, the IDO1 inhibitor BMS-986205 combined with the PD-1 antibody nivolumab produced only limited and transient relief of immunosuppression in the first-line treatment of advanced HCC and failed to translate into stable and reproducible population-level clinical benefit[165]. This outcome indicates that tumor-associated immunosuppression is typically sustained by multiple functionally redundant metabolic nodes, rendering single-pathway interventions highly susceptible to compensatory bypass mechanisms. Consequently, the central challenge of metabolic-immune combination therapy lies not in empirically intensifying inhibition at a single metabolic node but in precise patient stratification guided by intratumoral pharmacodynamic evidence. These challenges are further compounded by the fact that most HCC arises in the setting of cirrhosis. Patients frequently exist in a state of chronic systemic inflammation driven by cirrhosis-associated immune dysfunction (CAID) together with limited nutritional and metabolic reserves, such that immune function often approaches the lower threshold of efficacy[166,167]. Under these conditions, metabolic interventions that deviate from an optimal therapeutic window are more likely to preferentially impair antitumor immune responses and thereby constrain overall clinical benefit. An additional barrier is that the liver serves as a central organ for tumor immune regulation and a major site of cumulative treatment-related toxicity, which imposes hepatic reserve as a physiological constraint on the feasibility of combination therapies. To mitigate the competing risk of death from hepatic decompensation, most contemporary HCC clinical trials selectively enroll patients with preserved liver function to maximize detection of antitumor efficacy signals[168,169]. However, because most patients have chronic liver disease or cirrhosis, such selective enrollment inherently limits robust assessment of efficacy and safety in individuals with impaired hepatic function and restricts the generalizability of trial conclusions to real-world populations. Ultimately, potential hepatotoxicity introduced by combining metabolic agents with ICIs frequently compromises treatment feasibility, manifesting as dose reductions, premature discontinuation, hepatic decompensation, or early mortality[170,171]. Even in first-line systemic therapy for advanced HCC, early death is not uncommon and is strongly associated with baseline hepatic dysfunction, indicating that patients with insufficient liver reserve may undergo rapid decompensation before immunotherapy has sufficient time to confer clinical benefit[172]. Accordingly, any combination strategy that increases hepatic metabolic burden is likely to encounter a markedly narrowed safety window and limited clinical extrapolability. Future study designs should therefore place liver function-based stratification and liver disease-specific safety endpoints on equal priority with antitumor efficacy.
CONCLUSION
HCC orchestrates systemic rewiring of glucose, lipid, and amino acid metabolic networks that not only sustains the metabolic demands of continuous tumor cell proliferation but also profoundly reshapes the TME through nutrient competition, accumulation of metabolic byproducts, and modulation of key signaling pathways. Across multiple intersecting nodes, metabolic and immune programs engage in self-reinforcing amplification loops that stabilize immune evasion, thereby providing a conceptual framework for understanding the marked heterogeneity of immunotherapeutic responses observed in HCC. Metabolic-immune combination therapy may offer a strategy to overcome current therapeutic limitations and advance HCC management toward more precise and integrated treatment paradigms. However, substantial challenges remain, and translation into effective clinical application will require further investigation.
DECLARATIONS
Acknowledgments
All figures of the manuscript, including the graphical abstract, were created in BioRender. [Created in BioRender. Wang, Y. (2026) https://BioRender.com/5t0zh9m].
Authors’ contributions
Conceptualized the study, reviewed and edited the manuscript, acquired funding, and supervised the research: Zhou J, Yang XR
Conducted the investigation, prepared the original draft, and performed the visualization: Wang Y, Lu YJ
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was jointly supported by the National Natural Science Foundation of China (82072715, 82341027, and 82488101), the Noncommunicable Chronic Diseases-National Science and Technology Major Project (2024ZD0525400 and 2024ZD0525406), the National Ten-thousand Talent Program, the Shanghai Municipal Science and Technology Major Project, the Shanghai Science and Technology Commission (22S31901800, 24JS2840300, and 25JS2850400), the Shanghai Municipal Health Commission (2022LJ005), the Eastern Talent Program (Leading Project), and the Zhongshan Hospital Science Foundation (ZP2023-017).
Conflicts of interest
Yang XR is the Guest Editor of the Special Issue Advancing Multimodal Approaches in Liver Cancer: From Tumor Heterogeneity to Precision Therapies and an Associate Chief Editor of Hepatoma Research. Yang XR was not involved in any part of the editorial process for this manuscript, including reviewer selection, manuscript handling, or decision-making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Chan SL, Sun HC, Xu Y, et al. The Lancet Commission on addressing the global hepatocellular carcinoma burden: comprehensive strategies from prevention to treatment. Lancet. 2025;406:731-78.
2. Balogh J, Victor D 3rd, Asham EH, et al. Hepatocellular carcinoma: a review. J Hepatocell Carcinoma. 2016;3:41-53.
3. Wang Y, Wang P, Zhang Z, Zhou J, Fan J, Sun Y. Dissecting the tumor ecosystem of liver cancers in the single-cell era. Hepatol Commun. 2023;7:e0248.
4. Yan J, Jiang Z, Zhang S, et al. Spatial-temporal heterogeneities of liver cancer and the discovery of the invasive zone. Clin Transl Med. 2025;15:e70224.
5. Khan AA, Liu ZK, Xu X. Recent advances in immunotherapy for hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int. 2021;20:511-20.
6. Llovet JM, Castet F, Heikenwalder M, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19:151-72.
7. Li X, Zhou L, Xu X, et al. Metabolic reprogramming in hepatocellular carcinoma: a bibliometric and visualized study from 2011 to 2023. Front Pharmacol. 2024;15:1392241.
8. Yang F, Hilakivi-Clarke L, Shaha A, et al. Metabolic reprogramming and its clinical implication for liver cancer. Hepatology. 2023;78:1602-24.
9. Jiang K, Liu H, Chen X, et al. Reprogramming of glucose metabolism by nanocarriers to improve cancer immunotherapy: recent advances and applications. Int J Nanomedicine. 2025;20:4201-34.
10. Chen H, Wu Q, Peng L, et al. Mechanism, clinical significance, and treatment strategy of Warburg effect in hepatocellular carcinoma. J Nanomater. 2021;2021:1-10.
11. Shang RZ, Qu SB, Wang DS. Reprogramming of glucose metabolism in hepatocellular carcinoma: Progress and prospects. World J Gastroenterol. 2016;22:9933-43.
12. DeWaal D, Nogueira V, Terry AR, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. 2018;9:446.
13. He S, Tang S. WNT/β-catenin signaling in the development of liver cancers. Biomed Pharmacother. 2020;132:110851.
14. Park S, Hall MN. Metabolic reprogramming in hepatocellular carcinoma: mechanisms and therapeutic implications. Exp Mol Med. 2025;57:515-23.
15. Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008;38:2438-50.
16. Ho PC, Bihuniak JD, Macintyre AN, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162:1217-28.
17. Hu B, Yu M, Ma X, et al. IFNα potentiates anti-PD-1 efficacy by remodeling glucose metabolism in the hepatocellular carcinoma microenvironment. Cancer Discov. 2022;12:1718-41.
18. Yuan Y, Niu Y, Huang Z, et al. USP14-mediated metabolic competition impairs CD8+ T cell immunosurveillance in hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2025;122:e2510576122.
19. Lin SC, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab. 2018;27:299-313.
20. Chen G, Li MY, Yang JY, Zhou ZH. Will AMPK be a potential therapeutic target for hepatocellular carcinoma? Am J Cancer Res. 2024;14:3241-58.
21. Cho S, Kim W, Yoo D, et al. Impact of glucose metabolism on PD-L1 expression in sorafenib-resistant hepatocellular carcinoma cells. Sci Rep. 2024;14:1751.
22. Chen DP, Ning WR, Jiang ZZ, et al. Glycolytic activation of peritumoral monocytes fosters immune privilege via the PFKFB3-PD-L1 axis in human hepatocellular carcinoma. J Hepatol. 2019;71:333-43.
23. Lu LG, Zhou ZL, Wang XY, et al. PD-L1 blockade liberates intrinsic antitumourigenic properties of glycolytic macrophages in hepatocellular carcinoma. Gut. 2022;71:2551-60.
24. Patsoukis N, Bardhan K, Chatterjee P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692.
25. Davern M, Donlon NE, O’Connell F, et al. Nutrient deprivation and hypoxia alter T cell immune checkpoint expression: potential impact for immunotherapy. J Cancer Res Clin Oncol. 2023;149:5377-95.
26. Yang X, Liu Y, Wang P, et al. Targeting PDHK1 by DCA to restore NK cell function in hepatocellular carcinoma. Mol Cancer Ther. 2024;23:1731-42.
27. Estrella V, Chen T, Lloyd M, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013;73:1524-35.
28. Zhang J, Dong K, Zhang X, Li C, Yu J, Wang W. Characteristics of lactate metabolism phenotype in hepatocellular carcinoma. Sci Rep. 2023;13:19674.
29. Quinn WJ 3rd, Jiao J, TeSlaa T, et al. Lactate limits T cell proliferation via the NAD(H) redox state. Cell Rep. 2020;33:108500.
30. Fischer K, Hoffmann P, Voelkl S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812-9.
31. Liu S, Pan Y, Liu W, et al. Lactylation-driven MVP upregulation boosts immunotherapy resistance by inhibiting PD-L1 degradation in hepatocellular carcinoma. J Immunother Cancer. 2025;13:e012230.
32. Peralta RM, Xie B, Lontos K, et al. Dysfunction of exhausted T cells is enforced by MCT11-mediated lactate metabolism. Nat Immunol. 2024;25:2297-307.
33. Hui S, Ghergurovich JM, Morscher RJ, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551:115-8.
34. Zhang Q, Liu J, Lin H, Lin B, Zhu M, Li M. Glucose metabolism reprogramming promotes immune escape of hepatocellular carcinoma cells. Explor Target Antitumor Ther. 2023;4:519-36.
35. Ying S, Liu H, Zhang Y, Mei Y. Harnessing dendritic cell function in hepatocellular carcinoma: advances in immunotherapy and therapeutic strategies. Vaccines. 2025;13:496.
36. Shen J, Wu Z, Zhou Y, et al. Knockdown of SLC16A3 decreases extracellular lactate concentration in hepatocellular carcinoma, alleviates hypoxia and induces ferroptosis. Biochem Biophys Res Commun. 2024;733:150709.
37. Multhoff G, Vaupel P. Lactate-avid regulatory T cells: metabolic plasticity controls immunosuppression in tumour microenvironment. Signal Transduct Target Ther. 2021;6:171.
38. Gu J, Zhou J, Chen Q, et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-β signaling in regulatory T cells. Cell Rep. 2022;39:110986.
39. Colegio OR, Chu NQ, Szabo AL, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559-63.
40. Cai J, Zhang P, Cai Y, et al. Lactylation-driven NUPR1 promotes immunosuppression of tumor-infiltrating macrophages in hepatocellular carcinoma. Adv Sci. 2025;12:e2413095.
41. Han S, Bao X, Zou Y, et al. d-lactate modulates M2 tumor-associated macrophages and remodels immunosuppressive tumor microenvironment for hepatocellular carcinoma. Sci Adv. 2023;9:eadg2697.
42. Ramière C, Rodriguez J, Enache LS, Lotteau V, André P, Diaz O. Activity of hexokinase is increased by its interaction with hepatitis C virus protein NS5A. J Virol. 2014;88:3246-54.
43. Chen L, Lin X, Lei Y, et al. Aerobic glycolysis enhances HBx-initiated hepatocellular carcinogenesis via NF-κBp65/HK2 signalling. J Exp Clin Cancer Res. 2022;41:329.
44. Teng CF, Hsieh WC, Wu HC, et al. Hepatitis B virus Pre-S2 mutant induces aerobic glycolysis through mammalian target of rapamycin signal cascade. PLoS One. 2015;10:e0122373.
45. Borden ES, Jorgensen A, Natri HM, Hastings KT, Buetow KH, Wilson MA. HCV- and HBV-mediated liver cancer converge on similar transcriptomic landscapes and immune profiles. HGG Adv. 2025;6:100373.
46. Javanmard D, Najafi M, Babaei MR, et al. Investigation of CTNNB1 gene mutations and expression in hepatocellular carcinoma and cirrhosis in association with hepatitis B virus infection. Infect Agent Cancer. 2020;15:37.
47. Wang Y, Guan Y, Abbas AR, et al. Molecular subtypes of hepatocellular carcinoma linked to liver cell lineages and clinical outcomes of combination immunotherapy. Cell Rep Med. 2025;6:102473.
48. Dantzer C, Dif L, Vaché J, Basbous S, Billottet C, Moreau V. Specific features of ß-catenin-mutated hepatocellular carcinomas. Br J Cancer. 2024;131:1871-80.
49. Lee SK, Lim J, Jhun JY, et al. Landscape of T-cell exhaustion heterogeneity and HBV integration in virus-related HCC revealed by whole-exome, transcriptome, and single-cell sequencing. JHEP Rep. 2025;7:101518.
50. Guichard C, Amaddeo G, Imbeaud S, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694-8.
51. Granja SC, Longatto-Filho A, de Campos PB, et al. Non-alcoholic fatty liver disease-related hepatocellular carcinoma: immunohistochemical assessment of markers of cancer cell metabolism. Pathobiology. 2022;89:157-65.
52. Yasukawa K, Shimada S, Akiyama Y, et al. ACVR2A attenuation impacts lactate production and hyperglycolytic conditions attracting regulatory T cells in hepatocellular carcinoma. Cell Rep Med. 2025;6:102038.
53. Li M, Wang L, Cong L, et al. Spatial proteomics of immune microenvironment in nonalcoholic steatohepatitis-associated hepatocellular carcinoma. Hepatology. 2024;79:560-74.
54. Wu YL, Cappuyns S, Loh A, et al. Impact of underlying liver disease on unresectable hepatocellular carcinoma treated with immune checkpoint inhibitors. BJC Rep. 2024;2:8.
55. Yang K, Wang X, Song C, et al. The role of lipid metabolic reprogramming in tumor microenvironment. Theranostics. 2023;13:1774-808.
56. Cheng Y, He J, Zuo B, He Y. Role of lipid metabolism in hepatocellular carcinoma. Discov Oncol. 2024;15:206.
57. Sena LA, Denmeade SR. Fatty acid synthesis in prostate cancer: vulnerability or epiphenomenon? Cancer Res. 2021;81:4385-93.
58. Batchuluun B, Pinkosky SL, Steinberg GR. Lipogenesis inhibitors: therapeutic opportunities and challenges. Nat Rev Drug Discov. 2022;21:283-305.
59. Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. 2020;122:4-22.
60. Ren M, Xu H, Xia H, Tang Q, Bi F. Simultaneously targeting SOAT1 and CPT1A ameliorates hepatocellular carcinoma by disrupting lipid homeostasis. Cell Death Discov. 2021;7:125.
61. Liu Z, Liu W, Wang W, et al. CPT1A-mediated fatty acid oxidation confers cancer cell resistance to immune-mediated cytolytic killing. Proc Natl Acad Sci U S A. 2023;120:e2302878120.
62. Wang M, Han J, Xing H, et al. Dysregulated fatty acid metabolism in hepatocellular carcinoma. Hepat Oncol. 2016;3:241-51.
63. Manzo T, Prentice BM, Anderson KG, et al. Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J Exp Med. 2020;217:e20191920.
64. Liang J, Liao J, Chang R, et al. Riplet promotes lipid metabolism changes associated with CD8 T cell exhaustion and anti-PD-1 resistance in hepatocellular carcinoma. Sci Immunol. 2025;10:eado3485.
65. Qin Y, Huo F, Feng Z, et al. CD36 promotes iron accumulation and dysfunction in CD8+ T cells via the p38-CEBPB-TfR1 axis in early-stage hepatocellular carcinoma. Clin Mol Hepatol. 2025;31:960-80.
66. Hashemi M, Sabouni E, Rahmanian P, et al. Deciphering STAT3 signaling potential in hepatocellular carcinoma: tumorigenesis, treatment resistance, and pharmacological significance. Cell Mol Biol Lett. 2023;28:33.
67. Yang Y, Zheng B, Han Q, Zhang C, Tian Z, Zhang J. Targeting blockage of STAT3 inhibits hepatitis B virus-related hepatocellular carcinoma. Cancer Biol Ther. 2016;17:449-56.
68. Wang H, Franco F, Tsui YC, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol. 2020;21:298-308.
69. Dhar S, Sarkar T, Bose S, et al. FOXP3 transcriptionally activates fatty acid scavenger receptor CD36 in tumour-induced Treg cells. Immunology. 2025;174:296-309.
70. Li J, Chen J, Yang G, Zhang S, Li P, Ye L. CD36 as a therapeutic target in tumor microenvironment and lipid metabolism. Anticancer Agents Med Chem. 2025;25:447-59.
71. Yu W, Lei Q, Yang L, et al. Contradictory roles of lipid metabolism in immune response within the tumor microenvironment. J Hematol Oncol. 2021;14:187.
72. Tyurin VA, Cao W, Tyurina YY, Gabrilovich DI, Kagan VE. Mass-spectrometric characterization of peroxidized and hydrolyzed lipids in plasma and dendritic cells of tumor-bearing animals. Biochem Biophys Res Commun. 2011;413:149-53.
73. Tang W, Sun G, Ji GW, et al. Single-cell RNA-sequencing atlas reveals an FABP1-dependent immunosuppressive environment in hepatocellular carcinoma. J Immunother Cancer. 2023;11:e007030.
74. Luo S, Tang R, Jiang L, et al. Exosomal FABP5 drives HCC progression via macrophage lipid metabolism and immune microenvironment remodeling. Front Immunol. 2025;16:1644645.
75. Al-Khami AA, Zheng L, Del Valle L, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. 2017;6:e1344804.
76. Li C, Xiong L, Yang Y, et al. Sorafenib enhanced the function of myeloid-derived suppressor cells in hepatocellular carcinoma by facilitating PPARα-mediated fatty acid oxidation. Mol Cancer. 2025;24:34.
77. Mok EHK, Leung CON, Zhou L, et al. Caspase-3-induced activation of SREBP2 drives drug resistance via promotion of cholesterol biosynthesis in hepatocellular carcinoma. Cancer Res. 2022;82:3102-15.
78. Sohda T, Iwata K, Hirano G, et al. 3-Hydroxyl-3-methylglutaryl-coenzyme A reductase is up regulated in hepatocellular carcinoma associated with paraneoplastic hypercholesterolemia. Med Mol Morphol. 2013;46:239-42.
79. Long H, Guo X, Qiao S, Huang Q. Tumor LXR expression is a prognostic marker for patients with hepatocellular carcinoma. Pathol Oncol Res. 2018;24:339-44.
80. Xi B, Luo FZ, He B, et al. High nuclear ABCG1 expression is a poor predictor for hepatocellular carcinoma patient survival. Hepatobiliary Pancreat Dis Int. 2022;21:370-7.
81. Fu B, Meng W, Zhao H, et al. GRAM domain-containing protein 1A (GRAMD1A) promotes the expansion of hepatocellular carcinoma stem cell and hepatocellular carcinoma growth through STAT5. Sci Rep. 2016;6:31963.
82. Jiang Y, Sun A, Zhao Y, et al.; Chinese Human Proteome Project (CNHPP) Consortium. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature. 2019;567:257-61.
83. Ma X, Bi E, Lu Y, et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. 2019;30:143-56.e5.
84. Yang W, Bai Y, Xiong Y, et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature. 2016;531:651-5.
85. Li Z, Wang Y, Xing R, et al. Cholesterol efflux drives the generation of immunosuppressive macrophages to promote the progression of human hepatocellular carcinoma. Cancer Immunol Res. 2023;11:1400-13.
86. Cao D, Liu H. Dysregulated cholesterol regulatory genes in hepatocellular carcinoma. Eur J Med Res. 2023;28:580.
87. Li K, Shi H, Zhang B, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther. 2021;6:362.
88. Tavazoie MF, Pollack I, Tanqueco R, et al. LXR/ApoE activation restricts innate immune suppression in cancer. Cell. 2018;172:825-40.e18.
89. Xie Y, Sun R, Gao L, et al. Chronic activation of LXRα sensitizes mice to hepatocellular carcinoma. Hepatol Commun. 2022;6:1123-39.
90. Attal N, Sullivan MT, Girardi CA, Thompson KJ, McKillop IH. Fatty acid binding protein-4 promotes alcohol-dependent hepatosteatosis and hepatocellular carcinoma progression. Transl Oncol. 2021;14:100975.
91. He T, Tao B, Yi C, et al. 27-Hydroxycholesterol promotes metastasis by SULT2A1-dependent alteration in hepatocellular carcinoma. Cancer Sci. 2022;113:2575-89.
92. Arguello G, Balboa E, Arrese M, Zanlungo S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim Biophys Acta. 2015;1852:1765-78.
93. Fujiwara N, Nakagawa H, Enooku K, et al. CPT2 downregulation adapts HCC to lipid-rich environment and promotes carcinogenesis via acylcarnitine accumulation in obesity. Gut. 2018;67:1493-504.
94. Tian Y, Yang B, Qiu W, et al. ER-residential Nogo-B accelerates NAFLD-associated HCC mediated by metabolic reprogramming of oxLDL lipophagy. Nat Commun. 2019;10:3391.
95. Fu R, Xue W, Liang J, et al. SOAT1 regulates cholesterol metabolism to induce EMT in hepatocellular carcinoma. Cell Death Dis. 2024;15:325.
96. Pfister D, Núñez NG, Pinyol R, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592:450-6.
97. Zhang S, Xu H, Li M, et al. An etiology-stratified single-cell atlas identifies FABP4 as a prognostic marker for MASLD-related HCC. J Hepatol. 2026;84:920-32.
98. Murai H, Kodama T, Maesaka K, et al. Multiomics identifies the link between intratumor steatosis and the exhausted tumor immune microenvironment in hepatocellular carcinoma. Hepatology. 2023;77:77-91.
99. Lin Q, Lin Y, Huang Y, et al. Hepatitis B virus x protein upregulates SREBP2 to modulate autophagy in hepatocellular carcinoma. Cancer Med. 2025;14:e70916.
100. Na TY, Shin YK, Roh KJ, et al. Liver X receptor mediates hepatitis B virus X protein-induced lipogenesis in hepatitis B virus-associated hepatocellular carcinoma. Hepatology. 2009;49:1122-31.
101. Camus G, Herker E, Modi AA, et al. Diacylglycerol acyltransferase-1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J Biol Chem. 2013;288:9915-23.
102. Song G, Shi Y, Zhang M, et al. Global immune characterization of HBV/HCV-related hepatocellular carcinoma identifies macrophage and T-cell subsets associated with disease progression. Cell Discov. 2020;6:90.
104. Ma HY, Yamamoto G, Xu J, et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J Hepatol. 2020;72:946-59.
105. Liu X, Ren B, Ren J, Gu M, You L, Zhao Y. The significant role of amino acid metabolic reprogramming in cancer. Cell Commun Signal. 2024;22:380.
106. De Matteis S, Ragusa A, Marisi G, et al. Aberrant metabolism in hepatocellular carcinoma provides diagnostic and therapeutic opportunities. Oxid Med Cell Longev. 2018;2018:7512159.
107. Ye Y, Yu B, Wang H, Yi F. Glutamine metabolic reprogramming in hepatocellular carcinoma. Front Mol Biosci. 2023;10:1242059.
108. Dai W, Shen J, Yan J, et al. Glutamine synthetase limits β-catenin-mutated liver cancer growth by maintaining nitrogen homeostasis and suppressing mTORC1. J Clin Invest. 2022;132:e161408.
109. Jin H, Wang S, Zaal EA, et al. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. Elife. 2020;9:e56749.
110. Wang W, Guo MN, Li N, Pang DQ, Wu JH. Glutamine deprivation impairs function of infiltrating CD8+ T cells in hepatocellular carcinoma by inducing mitochondrial damage and apoptosis. World J Gastrointest Oncol. 2022;14:1124-40.
111. Leone RD, Zhao L, Englert JM, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366:1013-21.
112. Miao L, Lu C, Zhang B, et al. Advances in metabolic reprogramming of NK cells in the tumor microenvironment on the impact of NK therapy. J Transl Med. 2024;22:229.
113. Cheng K, Cai N, Zhu J, Yang X, Liang H, Zhang W. Tumor-associated macrophages in liver cancer: from mechanisms to therapy. Cancer Commun. 2022;42:1112-40.
114. Santarsiero A, Convertini P, Iacobazzi D, Infantino V, Todisco S. Metabolic crossroad between macrophages and cancer cells: overview of hepatocellular carcinoma. Biomedicines. 2024;12:2684.
115. Chen J, Sun HW, Wang RZ, et al. Glutamate promotes CCL2 expression to recruit tumor-associated macrophages by restraining EZH2-mediated histone methylation in hepatocellular carcinoma. Oncoimmunology. 2025;14:2497172.
116. Sun HW, Wu WC, Chen HT, et al. Glutamine deprivation promotes the generation and mobilization of MDSCs by enhancing expression of G-CSF and GM-CSF. Front Immunol. 2020;11:616367.
117. Oh MH, Sun IH, Zhao L, et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Invest. 2020;130:3865-84.
118. Yan J, Chen D, Ye Z, et al. Molecular mechanisms and therapeutic significance of Tryptophan Metabolism and signaling in cancer. Mol Cancer. 2024;23:241.
119. Liu M, Wang X, Wang L, et al. Targeting the IDO1 pathway in cancer: from bench to bedside. J Hematol Oncol. 2018;11:100.
120. Li S, Li L, Wu J, et al. TDO promotes hepatocellular carcinoma progression. Onco Targets Ther. 2020;13:5845-55.
121. Li S, Han X, Lyu N, et al. Mechanism and prognostic value of indoleamine 2,3-dioxygenase 1 expressed in hepatocellular carcinoma. Cancer Sci. 2018;109:3726-36.
122. Krishnamurthy S, Gilot D, Ahn SB, et al. Involvement of kynurenine pathway in hepatocellular carcinoma. Cancers. 2021;13:5180.
124. Litzenburger UM, Opitz CA, Sahm F, et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget. 2014;5:1038-51.
125. Wu Z, Yan L, Lin J, Ke K, Yang W. Constitutive TDO2 expression promotes liver cancer progression by an autocrine IL-6 signaling pathway. Cancer Cell Int. 2021;21:538.
126. Liu Y, Liang X, Dong W, et al. Tumor-repopulating cells induce PD-1 expression in CD8+ T cells by transferring kynurenine and AhR activation. Cancer Cell. 2018;33:480-94.e7.
127. Stone TW, Williams RO. Modulation of T cells by tryptophan metabolites in the kynurenine pathway. Trends Pharmacol Sci. 2023;44:442-56.
128. Song H, Park H, Kim YS, et al. L-kynurenine-induced apoptosis in human NK cells is mediated by reactive oxygen species. Int Immunopharmacol. 2011;11:932-8.
129. Jia H, Yang H, Xiong H, Luo KQ. NK cell exhaustion in the tumor microenvironment. Front Immunol. 2023;14:1303605.
130. Barroso A, Mahler JV, Fonseca-Castro PH, Quintana FJ. Therapeutic induction of tolerogenic dendritic cells via aryl hydrocarbon receptor signaling. Curr Opin Immunol. 2021;70:33-9.
131. Shin JH, Zhang L, Murillo-Sauca O, et al. Modulation of natural killer cell antitumor activity by the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2013;110:12391-6.
132. St Paul M, Saibil SD, Lien SC, et al. IL6 induces an IL22+ CD8+ T-cell subset with potent antitumor function. Cancer Immunol Res. 2020;8:321-33.
133. McAlpine JA, Lu HT, Wu KC, Knowles SK, Thomson JA. Down-regulation of argininosuccinate synthetase is associated with cisplatin resistance in hepatocellular carcinoma cell lines: implications for PEGylated arginine deiminase combination therapy. BMC Cancer. 2014;14:621.
134. Kim JS, Choi WM, Kim HI, et al. Synergistic effects of L-arginine and argininosuccinate synthetase 1 in inducing apoptosis in hepatocellular carcinoma. J Liver Cancer. 2025;25:79-90.
135. Zhang B, Dong LW, Tan YX, et al. Asparagine synthetase is an independent predictor of surgical survival and a potential therapeutic target in hepatocellular carcinoma. Br J Cancer. 2013;109:14-23.
136. Tao X, Zuo Q, Ruan H, et al. Argininosuccinate synthase 1 suppresses cancer cell invasion by inhibiting STAT3 pathway in hepatocellular carcinoma. Acta Biochim Biophys Sin. 2019;51:263-76.
137. Mossmann D, Müller C, Park S, et al. Arginine reprograms metabolism in liver cancer via RBM39. Cell. 2023;186:5068-83.e23.
138. Geiger R, Rieckmann JC, Wolf T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167:829-42.e13.
139. Chivite-Lacaba M, Justo I, Utrero-Rico A, et al. Delineation of monocytic and early-stage myeloid-derived suppressor cells in the peripheral blood of patients with hepatocarcinoma. Int J Cancer. 2025;156:2416-28.
140. Jeong JM, Choi SE, Shim YR, et al. CX3CR1+ macrophages interact with HSCs to promote HCC through CD8+ T-cell suppression. Hepatology. 2025;82:655-68.
141. Karadima E, Chavakis T, Alexaki VI. Arginine metabolism in myeloid cells in health and disease. Semin Immunopathol. 2025;47:11.
142. Casero RA Jr, Murray Stewart T, Pegg AE. Polyamine metabolism and cancer: treatments, challenges and opportunities. Nat Rev Cancer. 2018;18:681-95.
143. Puleston DJ, Buck MD, Klein Geltink RI, et al. Polyamines and eIF5A hypusination modulate mitochondrial respiration and macrophage activation. Cell Metab. 2019;30:352-63.e8.
144. Holbert CE, Casero RA Jr, Stewart TM. Polyamines: the pivotal amines in influencing the tumor microenvironment. Discov Oncol. 2024;15:173.
145. Mondanelli G, Bianchi R, Pallotta MT, et al. A relay pathway between arginine and tryptophan metabolism confers immunosuppressive properties on dendritic cells. Immunity. 2017;46:233-44.
146. Alexander ET, Minton A, Peters MC, Phanstiel O 4th, Gilmour SK. A novel polyamine blockade therapy activates an anti-tumor immune response. Oncotarget. 2017;8:84140-52.
147. Su H, Liu Y, Huang J. Ferroptosis-related gene SLC1A5 is a novel prognostic biomarker and correlates with immune microenvironment in HBV-related HCC. J Clin Med. 2023;12:1715.
148. Kuramitsu Y, Harada T, Takashima M, et al. Increased expression and phosphorylation of liver glutamine synthetase in well-differentiated hepatocellular carcinoma tissues from patients infected with hepatitis C virus. Electrophoresis. 2006;27:1651-8.
149. Ziki RA, Colnot S. Glutamine metabolism, a double agent combating or fuelling hepatocellular carcinoma. JHEP Rep. 2024;6:101077.
150. Lercher A, Popa AM, Viczenczova C, et al. Hepatocyte-intrinsic type I interferon signaling reprograms metabolism and reveals a novel compensatory mechanism of the tryptophan-kynurenine pathway in viral hepatitis. PLoS Pathog. 2020;16:e1008973.
151. Lercher A, Bhattacharya A, Popa AM, et al. Type I interferon signaling disrupts the hepatic urea cycle and alters systemic metabolism to suppress T cell function. Immunity. 2019;51:1074-87.e9.
152. Sun S, Li Y, Han S, Jia H, Li X, Li X. A comprehensive genome-wide profiling comparison between HBV and HCV infected hepatocellular carcinoma. BMC Med Genomics. 2019;12:147.
153. Du K, Chitneni SK, Suzuki A, et al. Increased glutaminolysis marks active scarring in nonalcoholic steatohepatitis progression. Cell Mol Gastroenterol Hepatol. 2020;10:1-21.
154. Teilhet C, Morvan D, Joubert-Zakeyh J, et al. Specificities of human hepatocellular carcinoma developed on non-alcoholic fatty liver disease in absence of cirrhosis revealed by tissue extracts ¹H-NMR spectroscopy. Metabolites. 2017;7:49.
155. Zhao X, Ren T, Li S, et al. A new perspective on the therapeutic potential of tumor metastasis: targeting the metabolic interactions between TAMs and tumor cells. Int J Biol Sci. 2024;20:5109-26.
156. Gao M, Huang A, Sun Z, et al. Granulocytic myeloid-derived suppressor cell population increases with the severity of alcoholic liver disease. J Cell Mol Med. 2019;23:2032-41.
157. De Chiara F, Heebøll S, Marrone G, et al. Urea cycle dysregulation in non-alcoholic fatty liver disease. J Hepatol. 2018;69:905-15.
158. Chen P, Geng H, Ma B, et al. Integrating spatial omics and single-cell mass spectrometry imaging reveals tumor-host metabolic interplay in hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2025;122:e2505789122.
159. Hartmann FJ. Spatial immunometabolism: integrating technologies to decode cellular metabolism in tissues. Eur J Immunol. 2025;55:e70094.
160. Liu Y, Dai Y, Wang L. Spatial omics at the forefront: emerging technologies, analytical innovations, and clinical applications. Cancer Cell. 2026;44:24-49.
161. Zhang Y, Shi M, Li M, Qin S, Miao D, Bai Y. Dynamic single-cell metabolomics reveals cell-cell interaction between tumor cells and macrophages. Nat Commun. 2025;16:4582.
162. Larson PEZ, Bernard JML, Bankson JA, et al.; HP 13C MRI Consensus Group. Current methods for hyperpolarized [1-13C]pyruvate MRI human studies. Magn Reson Med. 2024;91:2204-28.
163. Binan L, Jiang A, Danquah SA, et al. Simultaneous CRISPR screening and spatial transcriptomics reveal intracellular, intercellular, and functional transcriptional circuits. Cell. 2025;188:2141-58.e18.
164. Abou-Alfa GK, Qin S, Ryoo BY, et al. Phase III randomized study of second line ADI-PEG 20 plus best supportive care versus placebo plus best supportive care in patients with advanced hepatocellular carcinoma. Ann Oncol. 2018;29:1402-8.
165. Huynh JC, Cho M, Monjazeb A, et al. Phase I/II trial of BMS-986,205 and nivolumab as first line therapy in hepatocellular carcinoma. Invest New Drugs. 2024;42:35-43.
166. Tan SY, Kelkar Y, Hadjipanayis A, Shipstone A, Wynn TA, Hall JP. Metformin and 2-deoxyglucose collaboratively suppress human CD4+ T cell effector functions and activation-induced metabolic reprogramming. J Immunol. 2020;205:957-67.
167. Albillos A, Martin-Mateos R, Van der Merwe S, Wiest R, Jalan R, Álvarez-Mon M. Cirrhosis-associated immune dysfunction. Nat Rev Gastroenterol Hepatol. 2022;19:112-34.
168. Piñero F, da Fonseca LG. Trial eligibility in advanced hepatocellular carcinoma: Does it support clinical practice in underrepresented subgroups? World J Gastroenterol. 2021;27:3429-39.
169. Pasta A, Calabrese F, Jaffe A, et al. Safety and efficacy of atezolizumab/bevacizumab in patients with hepatocellular carcinoma and impaired liver function: a systematic review and meta-analysis. Liver Cancer. 2024;13:227-37.
170. Cheng AL, Qin S, Ikeda M, et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol. 2022;76:862-73.
171. da Fonseca LG, Piñero F, Anders M, et al. Immune-mediated adverse events following atezolizumab and bevacizumab in a multinational Latin American cohort of unresectable hepatocellular carcinoma. Oncotarget. 2025;16:348-60.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.