




Immune checkpoint inhibitors in hepatocellular carcinoma therapy: resistance mechanisms, liver transplantation challenges and management strategies
 0
0 
Abstract
Hepatocellular carcinoma (HCC) poses a significant clinical burden due to its aggressive nature, profound tumor heterogeneity, and limited therapeutic efficacy. While immune checkpoint inhibitors (ICIs) have revolutionized treatment paradigms and demonstrated considerable promise, the emergence of resistance mechanisms has posed a critical challenge in contemporary clinical oncology. The accelerated development of novel agents and innovative combination strategies has further complicated this resistance landscape. In this review, we present a unique and comprehensive analysis of ICI resistance mechanisms in HCC by integrating insights into primary resistance, acquired resistance, and host-related factors. Building upon this mechanistic framework, we explore emerging therapeutic strategies to overcome ICI resistance. Furthermore, we evaluate the dual role of ICIs in HCC management - serving as a neoadjuvant therapy for transplant candidates while simultaneously posing risks of post-transplant rejection. By bridging preclinical discoveries with clinical realities, this analysis aims to inform rational therapeutic design and optimize immuno-oncology trials for HCC patients.
Keywords
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer and the fourth leading cause of cancer-related mortality worldwide[1]. Although early-stage HCC can be clinically cured through surgical resection or traditional chemotherapy, the lack of apparent clinical symptoms in the early stages leads to more than 70% of patients being diagnosed at advanced stages, severely limiting therapeutic options[2]. The advent of immune checkpoint inhibitors (ICIs) has revolutionized therapeutic strategies for HCC. ICIs reactivate the antitumor immune response by blocking the interaction of inhibitory receptors on T cells with their ligands (e.g., PD-1, CTLA-4), thereby disarming the immune escape mechanism of tumor cells[3]. Recent studies have demonstrated the significant efficacy of ICIs in both immunotherapy[4] and neoadjuvant therapy[5] for HCC. However, the clinical response rate of ICIs is only 15%-30%, with persistent challenges of resistance[6]. Additionally, their application in liver transplant recipients faces risks of rejection, necessitating systematic exploration of molecular mechanisms and clinical translation strategies[5].
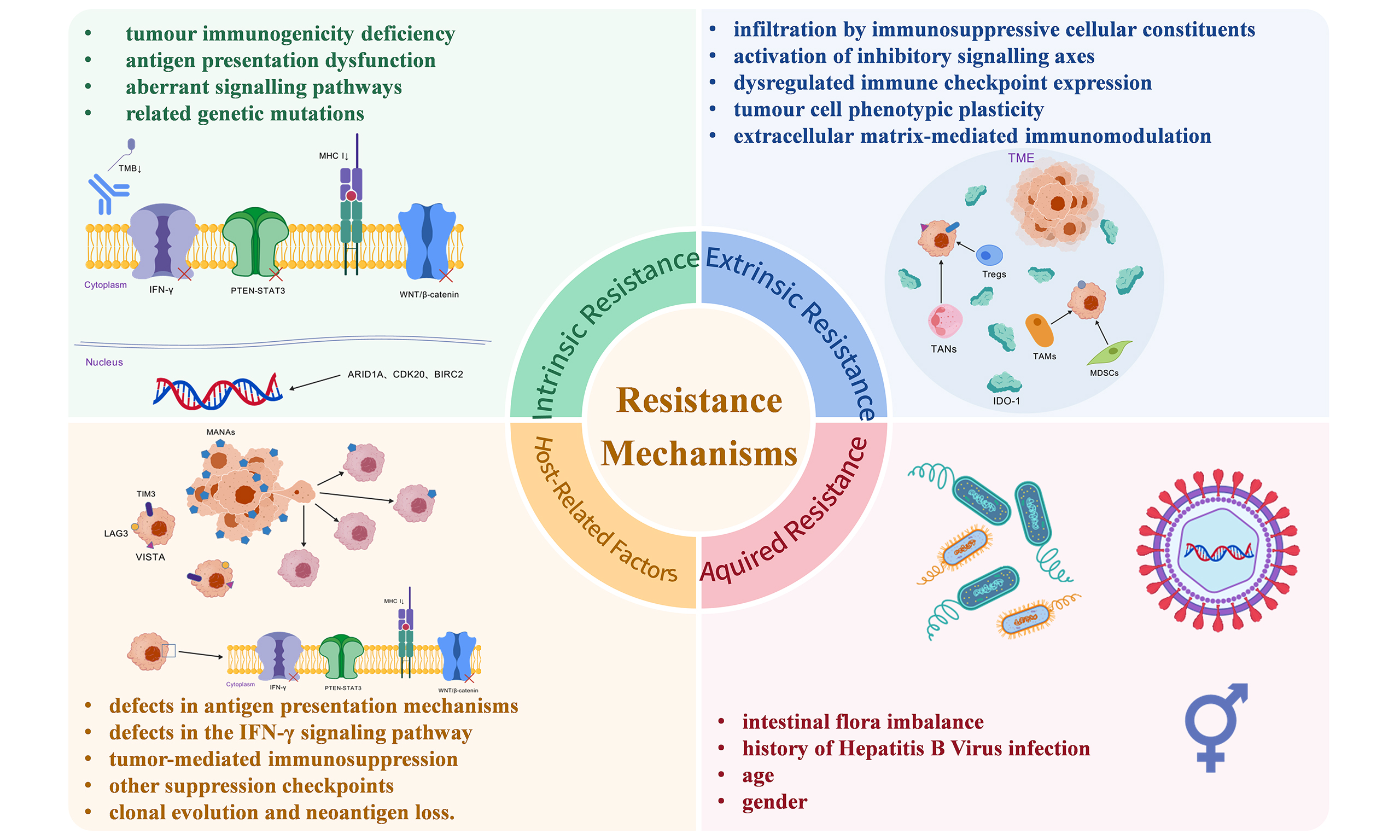
Recent studies have revealed the complex mechanisms underlying HCC resistance to ICIs, including primary resistance, acquired resistance, and host-related factors. Primary resistance can be categorized into intrinsic and extrinsic resistance. Intrinsic resistance mechanisms include tumor immunogenicity deficiency, antigen presentation dysfunction, aberrant signaling pathways, and related genetic mutations. Extrinsic resistance primarily arises from the immunosuppressive effects of the tumor microenvironment (TME)[7]. Acquired resistance is predominantly linked to neoantigen loss, immune cell exhaustion, and the upregulation of immune checkpoints[7]. Host-related factors are mainly involved in gut microbiota dysbiosis, chronic viral infections, and systemic immune dysregulation[8]. These mechanisms are intricately intertwined, suggesting that monotherapy strategies are insufficient to overcome therapeutic bottlenecks, necessitating the development of combination therapies based on multidimensional biomarkers.
Furthermore, the role of ICIs in liver transplant candidates represents a double-edged sword. On the one hand, as neoadjuvant therapy, ICIs may increase transplant feasibility through tumor downstaging; on the other hand, their immune-activating effects could increase posttransplant allograft rejection risk. Current evidence regarding the optimal timing of ICI administration (e.g., pretransplant washout period), indication selection (e.g., tumor biological characteristics), and immunosuppressive regimen adjustments remains contentious, necessitating the development of individualized strategies grounded in mechanistic studies[9,10].
This review aims to integrate fundamental research with clinical evidence, systematically dissect the multidimensional mechanisms of ICI resistance in HCC, and explore novel combination therapeutic approaches. Additionally, we critically assess the risk–benefit balance of ICIs during the perioperative period of liver transplantation (LT) to provide a theoretical framework for optimizing therapeutic decision making.
IMMUNOLOGICAL TME IN HCC
As a prototypical inflammation-associated malignancy, HCC features a tumor immune microenvironment (TIME) that orchestrates the dynamic equilibrium between immunogenicity and tolerance, crucially governing tumorigenesis, therapeutic response, and immunotherapy sensitivity[11]. The TIME in HCC demonstrates remarkable complexity and plasticity, characterized by (1) coexisting dual immunogenic/tolerogenic properties; (2) immunosuppression-dominated microenvironment homeostasis; (3) spatiotemporal heterogeneity in dynamic evolution; and (4) metabolic reprogramming-mediated immunomodulation[11].
Within the HCC TIME, immune cells exhibit a “double-edged sword” phenomenon: while executing antitumor functions through antigen presentation and cytotoxicity, they paradoxically facilitate immune evasion via checkpoint regulation and metabolic pathway manipulation[12]. CD8+ T cells mediate tumor killing via perforin, granzyme, and TNF-α secretion; however, their cytotoxic functions are markedly impaired under hypoxic/acidic conditions[13]. Notably, CD8+ T cell functional heterogeneity has prognostic significance: Notch1 signaling upregulation and exhausted phenotypes (PD-1+TIM-3+LAG-3+) correlate with immunotherapy resistance and poor outcomes[14], whereas the XCL1-positive subset predicts favorable survival in virus-associated HCC[15]. With regard to M1/M2 polarization dynamics, M1 macrophages exert antitumor effects via IL-12/TNF-α secretion, whereas TREM2-driven M2 polarization promotes HCC progression by suppressing CD8+ T cell infiltration and enhancing tumor glycolysis[16]. NK cells mediate tumor surveillance through IFN-γ/NF-α release, while hypoxic stress via HIF-1α-dependent mechanisms inhibits their activation and upregulates inhibitory receptors such as NKG2A[17]. Complementary regulatory networks involving B cells, tumor-associated neutrophils, and cytokine cascades further contribute to immune modulation[18].
The intrinsic immunosuppressive properties are particularly pronounced in HCC[19]. Tregs sustain immunosuppression via the TGF-β/IL-10 axis, and infiltration density is positively correlated with the stage of hepatoma[20]. MDSCs impair T cell and NK cell functions through the secretion of VEGF, arginase, and other mediators[21], while synergizing with Tregs to amplify immunosuppression[22]. CAFs establish physical/biochemical barriers through PD-L1/PD-L2 expression and FASL-mediated T cell apoptosis[23]. These cellular populations interact with hepatic sinusoidal endothelial cells, stellate cells, and tumor-derived exosomes to maintain an immune-tolerant ecosystem[24,25].
The heterogeneity of the TIME in HCC manifests in both temporal and spatial dimensions. Single-cell sequencing has revealed that early recurrent HCC harbors a distinct immune ecosystem characterized by expanded dendritic cell populations, reduced Tregs, and enriched CD8+ T cells in low-cytotoxicity states[26]. HCC is an “immune-cold tumor” phenotype characterized by inadequate T cell infiltration in tumor cores and stromal barrier formation in immune-excluded regions[27], potentially explaining suboptimal responses to ICIs.
Moreover, metabolic reprogramming constitutes a cornerstone of TIME modulation in HCC. Tumor cells generate lactate via aerobic glycolysis (the Warburg effect), which activates STAT3 signaling to upregulate PD-L1 expression and expand immunosuppressive Tregs/MDSCs while directly inhibiting CD8+ T cell oxidative phosphorylation[28]. TK1 catalyzes the production of the glycolytic metabolite dTMP and nonenzymatically interacts with PRMT1, establishing a glycolysis–methylation coupling network that sustains immunosuppressive metabolic homeostasis[29]. In lipid metabolism, CERS5 drives HCC progression through the sphingolipid–autophagy axis activation[30], whereas cholesterol dysregulation disrupts T cell receptor signaling via membrane lipid raft destabilization[28].
Collectively, these factors shape the unique immune evasion mechanisms of HCC and influence the clinical response and prognosis of patients receiving immunotherapy.
IMMUNE ESCAPE IN HCC
The immune escape mechanisms in HCC are categorized into the “3C” model: camouflage, coercion, and cytoprotection[31].
Malignancies employ antigenic camouflage as a critical immune evasion mechanism by subverting immune effector cell recognition, with the tumor mutational burden (TMB; somatic mutations/megabase) serving as a key determinant of neoantigen diversity and immunotherapy responsiveness[32]. Low-TMB tumors exhibit defective antigen processing/presentation machinery, which is correlated with ICI resistance and poor prognosis[33-35], whereas the suppression of immunogenic cell death (ICD) markers (e.g., ATP[36], annexins[37], calreticulin[38]) compromises APC recruitment/phagocytosis and T cell activation[39]. Notably, HCC-specific mechanisms involve HDAC8-mediated epigenetic silencing of the chemokine CCL4[40] and extracellular matrix (ECM) remodeling, which results in the establishment of immune exclusion zones[41,42]. When camouflage fails, tumors activate coinhibitory programs through PD-L1 upregulation[43,44], NKG2D ligand downregulation[45], and inactivation of the cGAS-STING pathway[46], coupled with metabolic immunosuppression via S-adenosylmethionine-induced T cell exhaustion[47]. Cytoprotective adaptations in HCC involve immune synapse destabilization through actin cytoskeletal remodeling[48], JAK-STAT hyperactivation, which confers resistance to IFN-γ-mediated apoptosis[49], and hypoxia-driven autophagy (HIF-1α-dependent), which degrades tumor antigens while expanding Tregs/MDSCs to sustain immunosuppressive niches[50].
Importantly, the unique immune evasion mechanisms of HCC are shaped by its viral oncogenic context and molecular crosstalk. Chronic HBV/HCV infection drives PD-L1 upregulation via PI3K-AKT-mTOR activation while suppressing MHC-I antigen presentation through HBx-mediated ERAP1 inhibition, which is compounded by viral genome integration-induced low TMB that dampens neoantigen immunogenicity[51]. Studies have revealed that the TGF-β signaling-upregulated noncoding RNA HDAC2-AS2 in HBV+HCC targets CDK9 in CD8+ T cells via exosomes, compromising their function and offering a novel therapeutic target for HBV-associated HCC[52]. Additionally, tumor-initiating cells (TICs) in HCC drive immune escape through unique interactions with neutrophils. CD49f-high TICs recruit neutrophils and establish an immunosuppressive milieu via the CXCL2-CXCR2 axis, evading CD8+ T cell attack. Neutrophils also secrete CCL4 to induce the phenotypic conversion of neighboring tumor cells toward TIC-like states, facilitating immune evasion. CD155 overexpression is central to these mechanisms and represents a potential therapeutic target against TICs[53]. Furthermore, HCC metabolic reprogramming is tightly coupled with immunosuppression. For example, S100A10 activates the cPLA2/5-LOX axis to initiate lipid metabolic reprogramming, elevating LTB4 levels and promoting CD8+ T cell exhaustion in HCC tissues, thereby driving immune escape and enhancing tumor growth and migration[54]. Similarly, S100A9 + MDSCs activate the ERK/NF-κB pathway in HCC cells, creating a self-sustaining “ETV5-S100A9-ERK/
HCC also exhibits distinct immunological features across various stages of tumor progression. Early recurrent HCC has unique immune escape characteristics, with increased proportions of CD161+CD8+ T cells in the microenvironment. These cells exhibit innate-like immunity, low cytotoxicity, and defects in clonal expansion. Concurrently, recurrent tumor cells competitively bind PD-L1 to CD80 on APCs, blocking CD28 costimulatory signaling and preventing T cell activation by APCs[26]. Additionally, studies indicate that immunosuppression in HCC progresses gradually and peaks at TNM stage II, unlike in most solid tumors, where it occurs early in carcinogenesis or before metastasis. Partial immune restoration is observed in TNM stage III tumors and is closely associated with increased neoantigen production[58].
The HCC immune checkpoint landscape is dominated by CTLA-4, PD-1/PD-L1, and LAG-3, which mechanistically demarcate T cell regulation across distinct phases. CTLA-4 antagonizes CD28 costimulation during priming by competitively binding with high affinity to B7-1/B7-2 on APCs, while PD-1 mediates suppression during the effector phase by engaging PD-L1/PD-L2 in chronic, antigen-rich microenvironments[59]. LAG-3 complements this suppression by recognizing MHC class II on APCs, inducing ITIM/ITSM domain phosphorylation and thereby exacerbating T cell exhaustion[60].
These multilayered evasion strategies enable HCC cells to orchestrate immune tolerance through the dual upregulation of ligands and the recruitment of stromal cells, creating an immunosuppressive niche. Precision-engineered ICIs counteract this tolerance by sterically blocking checkpoint axes, thereby reversing T cell anergy and restoring their cytotoxic potential against tumor cells.
MECHANISMS OF RESISTANCE TO ICIS IN HCCS
Despite the transformative potential of ICIs in HCC management, ICI resistance remains a multifaceted clinical challenge, mechanistically stratified into primary resistance, acquired resistance, and host-related factors [Table 1]. The following paragraphs highlight this topic.
Mechanisms of ICI resistance in HCC
| Mechanism | Effect | |
| Intrinsic resistance | TMB, MSI ↓ | Immune cell infiltration ↓[63-65] |
| IRGQ | Antigen presentation via MHC-I ↓[67] | |
| FASN | MHC-I expression ↓[71,72] | |
| WNT/β-catenin signaling ↑ | CCL5, Batf3 ↓, DC recruitment to TME ↓, antigen presentation ↓[73,74] | |
| NKG2D ↓, NK cell immune surveillance ↓[75] | ||
| JAK1/2 mutation | T cell infiltration and IFN-γ signaling ↓[79] | |
| PTEN-STAT3 mutation | Cytotoxic T cell killing of tumor cells ↓[80] | |
| MerTK | Ferroptosis ↓, immunosuppressive TME ↓[81] | |
| MYC ↑ | Immune escape ↑[82] | |
| TP53 mutation | Immune cell infiltration and function ↓[83] | |
| ARID1A mutation | IFN-γ signaling ↓[85] | |
| CDK20 ↑ | MDSCs ↑[86] | |
| BIRC2 ↑ | CD4+ T cells and CD8+ T cells ↓[87] | |
| Extrinsic resistance | MDSCs ↑ | NK cells ↓[89,90] |
| TANs ↑ | CD8+ T cells ↓[91] | |
| TAMs ↑ | T cells ↓[92,93] | |
| WNT/β-catenin signaling ↑ | CCL5 ↓, DC ↓, NK cells ↓[76] | |
| VEGF ↑ | Induces FasL-mediated immune resistance[94] | |
| TGF-β ↑ | CTLs and NK cells ↓, TAMs ↑[95] | |
| EMT | Induces tolerance[99,100] | |
| IDO-1 | Alters collagen matrix composition, abnormal collagen deposition ↑[102] | |
| Tryptophan and kynurenine ↑[103] | ||
| Acquired resistance | B2M mutation | MHC-I ↓[105-108] |
| PTEN | IFN-γ ↓[109] | |
| MANAs ↓ | TMB ↓[112] | |
| Host-related factors | Altered intestinal flora | Disturbed bile acid metabolism[115,116] |
| HBV history | Tregs, MDSCs, TAMs ↑, formation of “cold tumors”[118] |
Primary resistance
Intrinsic resistance
Intrinsic resistance mechanisms primarily involve tumor immunogenicity deficiency, dysfunctional antigen presentation, aberrant signaling pathways, and related genetic mutations.
Tumor immunogenicity is governed by the TMB and neoantigen diversity, where somatic mutations generate immunogenic neopeptides that are devoid of autoimmune risk[61]. The TMB, a metric that reflects the cancer mutation load, is positively correlated with neoantigen abundance and enhances T cell activation and immunogenicity. The TMB also serves as a biomarker for predicting the efficacy of PD-1 inhibitors[62]. Microsatellite instability (MSI) is associated with high TMB; however, most HCC patients exhibit low TMB and MSI levels[63]. Studies have revealed that HCC with a high TMB generates more neoantigens, activating antigen presentation and T cell responses to form an “immune-hot tumor” phenotype, characterized by increased infiltration of dendritic cells, Tregs, memory B cells, and CD8+ T cells[64]. In high-TMB HCC patients, the response to ICI treatment and prognosis are superior to those in low-TMB patients[65]. However, some studies have drawn conflicting conclusions: despite low TMB in HCC patients, no significant correlation exists between the TMB and prognosis[66], suggesting that the TMB may be merely one factor influencing ICI resistance and cannot reliably predict ICI efficacy in HCC.
Antigen presentation dysfunction represents a pivotal mechanism underlying primary resistance to ICIs in HCC and is predominantly mediated through MHC-I downregulation. IRGQ functions as a novel autophagy receptor that interacts with GABARAPL2 and LC3B and undergoes autophagy-dependent lysosomal trafficking to degrade misfolded MHC-I heavy chains, thereby evading CD8+ T cell surveillance. IRGQ overexpression in HCC models potently suppresses MHC-I-mediated antigen presentation[67]. Clinically, MHC-I expression inversely correlates with tumor progression (reduced in 35% of early-stage HCC patients)[68], whereas preserved MHC-I expression is associated with improved recurrence-free survival[69] and a reduced tumor burden[70]. Mechanistically, FASN regulates MHC-I stability and membrane localization via palmitoylation, a posttranslational modification critical for immunomodulatory proteins such as PD-L1[71,72].
Dysregulated oncogenic signaling critically underpins primary immune ICI resistance in HCC, wherein WNT/β-catenin activation suppresses CCL5 transcription and Batf3 depletion[73,74], impairing DC recruitment and antigen presentation while downregulating NKG2D ligands to compromise NK cell surveillance[75,76]. Clinical evidence shows that HCC patients with β-catenin activation exhibit significantly reduced response rates to PD-1 inhibitors[77,78]. Concurrently, JAK1/2 inactivation disrupts IFN-γ signaling, reducing T cell infiltration[79], whereas PTEN-STAT3 dysregulation promotes STAT3 nuclear translocation (prevalent in 60% of HCC patients), which inhibits T cell cytotoxicity and is correlated with poor prognosis[80]. MerTK orchestrates dual resistance mechanisms: ERK/SP1-mediated SLC7A11 activation suppresses ferroptosis, whereas MDSC recruitment via chemokine signaling cripples CD8+ T cell function, synergistically conferring resistance to PD-1/PD-L1 inhibitors[81].
Oncogenic mutations in HCC orchestrate intrinsic ICI resistance through multifaceted immune evasion mechanisms. MYC amplification (50%-70% of HCC patients) drives PD-L1 overexpression and immunosuppression[82], whereas TP53 mutation (~40% of HCC patients) impairs tumor suppressor function, recruits immunosuppressive cells, and remodels the TME immune landscape[83]. ARID1A mutations exhibit a paradoxical duality: enhancing TMB and PD-L1 expression via mismatch repair defects[84] while restricting IFN-γ responses through chromatin remodeling[85]. Moreover, CDK20 activation recruits MDSCs to suppress autologous CD8+ T cells[86], whereas BIRC2 overexpression inversely correlates with CD8+/CD4+ T cell infiltration, predicting reduced survival and ICI resistance[87].
Extrinsic resistance
Extrinsic resistance in HCC is predominantly orchestrated by the immunosuppressive TME, which includes infiltration by immunosuppressive cellular constituents, activation of inhibitory signaling axes, dysregulated immune checkpoint expression, tumor cell phenotypic plasticity, and ECM-mediated immunomodulation.
The TME harbors immunosuppressive cells such as Tregs, MDSCs, TANs, and tumor-associated macrophages (TAMs). While Tregs physiologically maintain immune homeostasis through T cell suppression and cytokine modulation, their pathological accumulation in HCC paradoxically impairs antitumor immunity[88]. MDSC infiltration is correlated with ICI resistance, resulting in immunosuppression via cysteine depletion, arginase-1/inducible nitric oxide synthase upregulation, reactive oxygen species generation, and Treg expansion[89]. Clinical cohorts have demonstrated that elevated monocyte-derived MDSCs in HCC patients inhibit NK cell activity[90]. CRKL overexpression drives TAN recruitment through β-catenin stabilization and VEGFα/CXCL1 upregulation, with PD-L1+ TAN subsets suppressing CD8+ T cells via ROS/anti-inflammatory factor secretion[91]. M2-polarized TAMs potentiate HCC progression and ICI resistance through protumorigenic secretomes and T cell suppression[92], whereas TAM-derived extracellular vesicles deliver the circPETH-encoded circPETH-147aa protein, which enhances tumor glycolysis via PKM2-mediated ALDOA-S36 phosphorylation and induces CD8+ T cell exhaustion through HuR-dependent methionine/leucine deprivation[93].
Immunosuppressive signaling axis activation in HCC involves a mechanistic overlap with intrinsic resistance pathways, notably through Wnt/β-catenin-mediated CCL5 suppression, DC exclusion, and NKG2D ligand downregulation, collectively impairing NK cell functionality[76]. Additionally, the VEGF signaling pathway regulates neovascularization in HCC and induces immune resistance by increasing FasL expression, triggering the apoptosis of tumor-infiltrating CD8+ T cells[94]. TGF-β signaling orchestrates dual immunosuppression: inhibiting CTL/NK cytotoxicity through IFN-γ blockade and NKG2D/NKp30 surface depletion while polarizing myeloid cells toward M2-TAM phenotypes and suppressing macrophage/DC/neutrophil maturation[95].
HCC exhibits compensatory upregulation of coinhibitory checkpoints beyond PD-1/PD-L1, with TIM-3 demonstrating DHHC9-mediated palmitoylation that stabilizes membrane localization on tumor-infiltrating CD8+ T/NK cells, driving exhaustion and correlating with poor prognosis[96]. The binding of TIM-3 to its ligand galectin-9 activates a complex signaling cascade that ultimately induces T cell exhaustion[97]. LAG-3 undergoes nondegradative ubiquitination by c-Cbl/Cbl-b E3 ligases upon activation, exposing the cryptic FSALE motif through basic residue-rich sequence–phospholipid interaction disruption to transmit immunosuppressive signals[98]. These checkpoints synergistically orchestrate PD-1/PD-L1 blockade resistance via combined T/NK cell exhaustion pathways.
Within the TME of HCC, tumor cells undergo adaptive epithelial–mesenchymal transition (EMT), driving ICI resistance through mechanisms centered on reshaping the TME into an immunosuppressive state and enhancing tumor invasiveness and adaptability. EMT promotes immunosuppressive TME formation by recruiting APCs, inducing tolerance, upregulating immune checkpoints, and resisting NK cell-mediated lysis[99,100].
Additionally, dense collagen matrices within the ECM impede T cell infiltration. Indoleamine 2,3-dioxygenase 1 (IDO-1), a heme-containing enzyme in the ECM, disrupts immune clearance. In HCC, IDO-1 exacerbates the immunosuppressive microenvironment by modulating dense collagen matrices through multiple mechanisms[101]. First, IDO-1 regulates TAMs and Tregs to alter collagen synthesis/degradation, modifying the ECM composition and structure to create a TME conducive to HCC progression. Second, IDO-1 suppresses T cell and NK cell functions, enabling immune evasion. Tumor cells then secrete factors to stimulate fibroblast-mediated collagen synthesis, increasing matrix density. Moreover, IDO-1 interacts with the TGF-β and VEGF pathways to regulate collagen metabolism. TGF-β promotes collagen synthesis[102], and IDO-1, which is regulated by TGF-β, synergistically enhances pathological collagen deposition. VEGF remodels ECM components during angiogenesis, collaborating with IDO-1 to foster a tumor-promoting niche. IDO-1 also induces tryptophan depletion and kynurenine accumulation in the TME, driving tumor resistance to ICIs[103].
Acquired resistance
Unlike primary resistance, acquired resistance refers to a patient who initially develops a clinical response to ICI therapy but subsequently experiences disease progression during treatment, characterized by dynamic post-treatment evolution. Since the mechanisms of acquired resistance in HCC have been poorly studied, we also report the mechanisms of acquired resistance identified in other malignancies. The mechanisms of acquired resistance in ICIs can be broadly classified into the following categories[104]: (1) defects in antigen-presentation mechanisms; (2) defects in the IFN-γ signaling pathway; (3) tumor-mediated immunosuppression; (4) other inhibitory checkpoints; and (5) clonal evolution and neoantigen loss.
Shared mechanisms underlie both primary and acquired ICI resistance, with a pathogenic convergence of β-2-microglobulin (B2M) loss-of-function mutations that destabilize MHC-I antigen presentation, a phenomenon recurrently observed in melanoma and lung cancer with acquired resistance[105-108]. Effector T cell-derived IFN-γ activates tumoricidal JAK-STAT signaling to upregulate MHC-I/PD-L1, although acquired resistance emerges via JAK1/JAK2 inactivation in melanoma models[105]. Concurrently, PTEN deletion drives PI3K-mediated immunosuppression through cytokine dysregulation and T cell exclusion[109], whereas WNT-β-catenin activation similarly promotes resistance via DC dysfunction, Treg expansion, and attenuated T cell infiltration in melanoma[110]. Malignancies further exhibit compensatory upregulation of alternative checkpoints (TIM3, LAG3, and VISTA) as a pathoadaptive response to sustained ICI pressure[107,111].
Clonal evolution and neoantigen loss represent unique mechanisms of acquired resistance. During immune editing, the immune system preferentially eliminates tumor cell clones with increased immunogenicity. Surviving tumor cells exhibit lower immunogenicity and lack critical neoantigen expression, which enables immune evasion during therapy and drives ICI resistance. Notably, NSCLC patients with post-ICI relapse exhibit loss of mutations encoding putative mutation-associated neoantigens (MANAs) in resistant tumor clones. Whole-exome sequencing revealed the disappearance of 7-18 MANAs in resistant clones that were present in pretreatment tumors[112]. Additionally, clonal evolution may generate intratumor heterogeneity (ITH)[113], resulting in diminished tumor immunogenicity. Studies of multifocal HCC reveal mutational and copy number variation heterogeneity across lesions, with complex ITH and clonal evolution patterns impairing ICI efficacy[114].
Although abnormalities in various immune cells are commonly observed across different types of drug-resistant environments, they play significantly distinct roles in the classification and mechanisms of drug resistance. In intrinsic resistance, they constitute pre-existing potent inhibitory barriers[67,68,71,72]. In extrinsic resistance, they serve as core executors of the TME by providing active protection and survival support[99,100]. In acquired resistance, they are key factors in the dynamic co-evolution with tumor cells, participating in immune editing and the remodeling of adaptive immune suppression[105-108].
Host-related factors
The gut microbiota modulates HCC responsiveness and resistance to ICIs. Dysbiosis disrupts bile acid metabolism (e.g., deoxycholic acid accumulation), impairing intrahepatic antitumor NKT cell recruitment[115,116]. Furthermore, post-ICI T cell responses correlate with the abundance of Bacteroides in the gut. CTLA-4 blockade induces mucosal colonization of Bacteroides species (e.g., B. fragilis), triggering TH1 cell hyperactivation and IL-12-dependent immune responses that enhance DC antigen presentation and proinflammatory cytokine production, thereby modulating ICI efficacy[117]. Most HCC patients have a history of HBV infection, where chronic inflammation expands Tregs, MDSCs, and M2 macrophages, suppresses effector T cell function, and fosters an “immune-cold tumor” phenotype[118]. Additionally, demographic variables, including age and sex, differentially regulate antitumor immunity through endocrine–immunologic crosstalk, potentially contributing to disparities in therapeutic outcomes[119,120].
DILEMMA OF ICIS IN LT
Currently, LT remains the most effective treatment for HCC, yet the application of ICIs in LT presents significant dilemmas and challenges.
Contradiction between LT and ICIs
Pre-LT ICI administration presents a clinical conundrum: tumor control to preserve transplant eligibility vs. acute rejection after liver transplantation (LT-AR) risk. A case series of 10 PD-1 inhibitor-treated LT candidates demonstrated acute rejection (30%) manifesting as lobular necrosis requiring retransplantation[121], with the treatment-transplantation interval emerging as a critical determinant - washout periods > 50 days reduced rejection rates to 16% (non-ICI historical baseline) without compromising oncologic outcomes[122]. These findings provide critical evidence for establishing a safe clinical window.
Similarly, post-LT use of ICIs in patients with HCC recurrence helps control tumor progression but risks disrupting immune tolerance, increasing the likelihood of graft rejection. Such rejection may shorten overall survival (OS) and progression-free survival (PFS)[123]. Studies indicate that 25% of LT recipients treated with ICIs develop acute rejection, with the rejection risk escalating when ICI initiation is closer to transplantation[124]. Furthermore, PD-1 inhibitor-treated LT recipients exhibit higher rejection rates than those receiving anti-CTLA-4 agents, suggesting differential rejection risks across ICI classes[123]. Moreover, the long-term immunosuppressants (e.g., tacrolimus and cyclosporine) required post-LT to prevent rejection may impair ICI antitumor efficacy by suppressing T cell function and increasing the risk of recurrence[123]. The immunosuppressive state also promotes the enrichment of immunosuppressive cells (e.g., Tregs and TAMs) in the TME, exacerbating therapeutic resistance[9]. The clinical data concerning the pre- and postoperative application of ICIs in LT patients are presented in Table 2.
Clinical data on pre- and postoperative application of ICIs in LT patients
| Research type | N | ICIs before/after LT | ICIs before/after LT | Safety |
| Retrospective cohort study[125] | 83 | Before | Camrelizumab (37.3%), Pembrolizumab (21.7%), Sintilimab (16.9%), Tislelizumab (13.3%), Nivolumab (6%), Atezolizumab (4.8%) | LT-AR (27.7%): fully recovered (65.2%), partially improved (8.7%), died (26.1%) |
| Retrospective cohort study[126] | 6 | Before | Atezolizumab + Bevacizumab (66.7%), Nivolumab + Ipilimumab (16.7%), Nivolumab (16.7%) | No patients had clinical evidence of rejection |
| Retrospective cohort study[121] | 10 | Before | Pembrolizumab or Camrelizumab + Lenvatinib | LT-AR (30%): died (66.7%) |
| Meta-analysis | 91 | Before | Nivolumab (49.5%), Pembrolizumab (23.1%), Atezolizumab + Bevacizumab (15.4%), Sintilimab (6.6%), Camrelizumab (4.4%), Durvalumab (1%) | LT-AR (26.4%): partially improved (83.3%), died (8.3%) |
| Pooled analysis | 52 | After | PD-1 blockade, Nivolumab, Pembrolizumab, Cemiplimab, Ipilimumab, Atezolizumab + Bevacizumab | LT-AR (28.8%): died (46.7%) |
Recurrence of HCC after LT
Standard post-LT immunosuppressive regimens induce an immunotolerant niche in recurrent HCC, characterized by Treg enrichment, TAM polarization, and tertiary lymphoid structure (TLS) depletion. These agents promote Treg expansion by suppressing the CD28/CD80 axis while facilitating TAM accumulation by impairing T cell-mediated macrophage regulation. Treg/TAM-derived IL-10 and TGF-β synergistically inhibit CD8+ T cell cytotoxicity and upregulate PD-L1, thereby establishing an ICI-refractory microenvironment[127]. Concurrently, mTOR pathway activation drives TLS loss through cell cycle dysregulation, impairing antitumor immunity and reducing ICI response rates by 42% compared with those of TLS-positive counterparts[128]. This immunosuppressive landscape underlies the 15% post-LT HCC recurrence rate, with ICIs demonstrating limited efficacy (partial response rates < 20%) owing to adaptive TME remodeling and host immune reconstitution failure[129]. Studies have shown that only a subset of LT recipients with recurrent HCC achieve a partial response to nivolumab, while most exhibit treatment resistance, likely due to an altered TME and host immune adaptations [Figure 1].
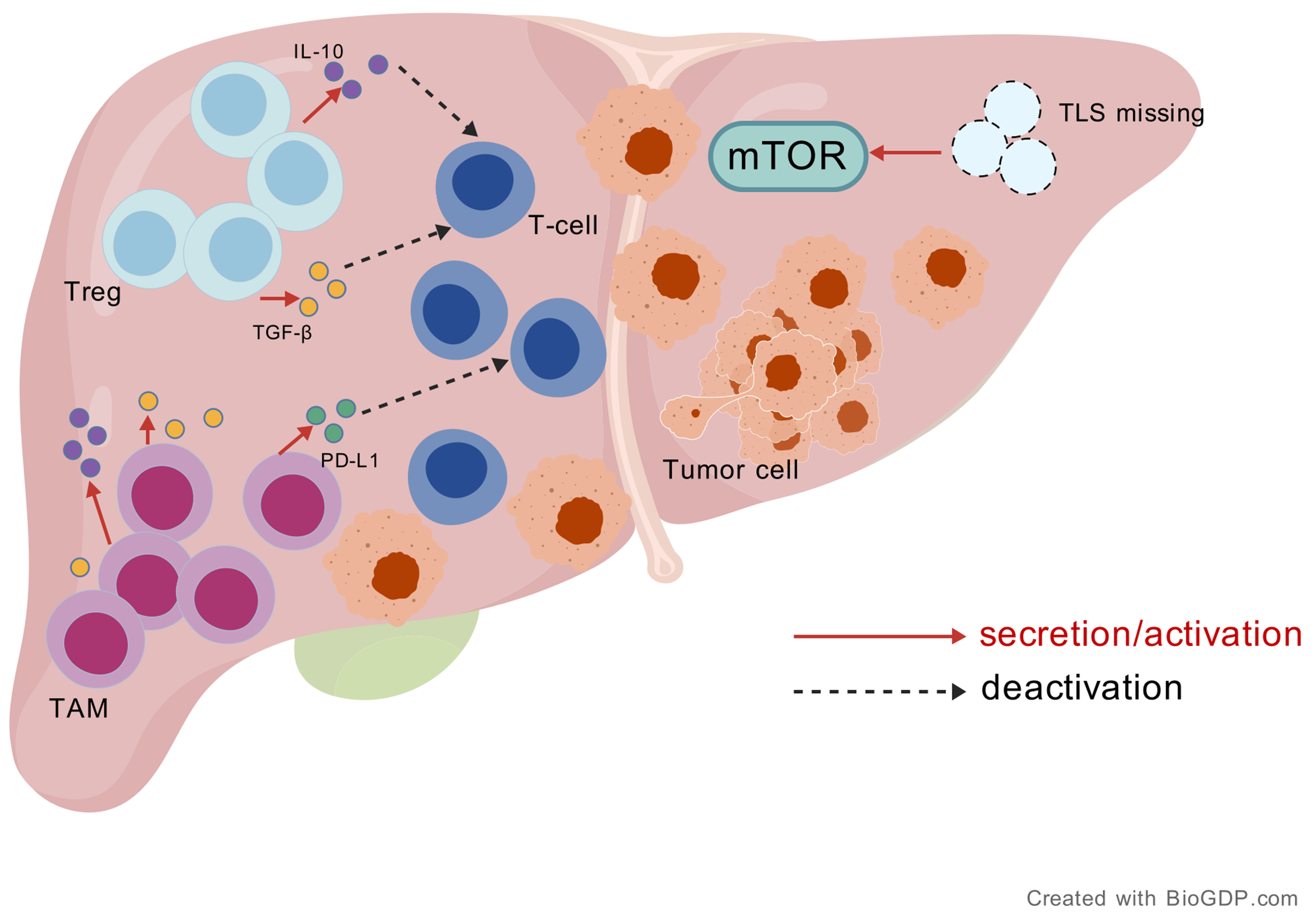
Figure 1. Schematic diagram of the TME associated with the recurrence of HCC after LT (created with BioGDP.com). TME: Tumor microenvironment; HCC: hepatocellular carcinoma; LT: liver transplantation.
Clinical treatment dilemmas and corresponding strategies
Post-LT HCC patients face multiple clinical challenges, including the lack of reliable biomarkers to predict ICI efficacy, with existing markers (e.g., PD-L1 expression, TMB) showing limited applicability under complex immunosuppressive conditions. Emerging radiomic-based quantification of TLS abundance shows promise as a noninvasive prognostic indicator for HCC-LT outcomes[128], although multicenter validation is needed. The exclusion of transplant recipients from pivotal ICI trials exacerbates evidence gaps, necessitating individualized strategies such as optimized immunosuppressant titration (e.g., mTOR inhibitors such as sirolimus)[128], extended pre-LT ICI washout (> 50 days)[122], and graft PD-L1 profiling to guide PD-1/PD-L1 antibody selection, and PD-L1-negative grafts exhibit reduced rejection risk[130]. Thus, for LT recipients with recurrence refractory to other therapies, PD-L1 testing of the graft is recommended. PD-L1-negative patients may receive PD-1/PD-L1 monoclonal antibodies as salvage therapy, with close monitoring of liver function and vigilance for acute rejection. Furthermore, novel biomarkers such as TLS abundance hold promise for refining patient stratification and advancing precision therapy[128]. Specifically, the management process for liver transplant candidates using ICIs in clinical practice is as described in Figure 2.
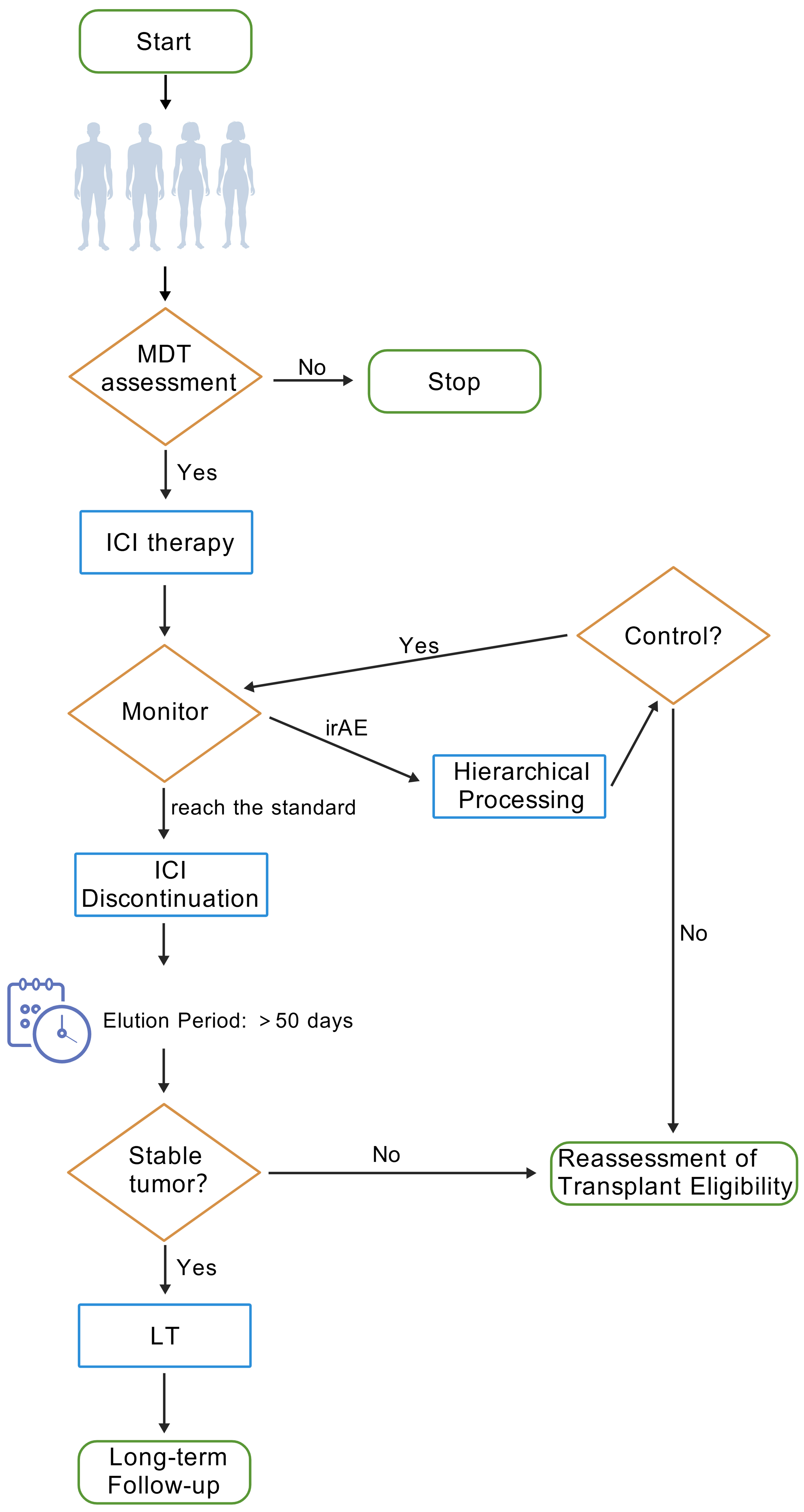
Figure 2. A clinical management flowchart for ICI use in LT candidates (created with BioGDP.com). ICI: Immune checkpoint inhibitor; LT: liver transplantation.
ICI resistance in post-LT HCC arises from the interplay of immunosuppression, TME remodeling, and therapeutic strategy limitations. Future efforts should focus on clinical trials targeting transplant populations to develop biomarker-guided precision therapies and novel combinations while optimizing immune management to balance graft preservation and antitumor efficacy.
TREATMENT STRATEGIES FOR HCC RESISTANT TO ICIS
Combination therapy
Locoregional Therapy combined with ICIs
Locoregional therapies (LRTs), such as transarterial chemoembolization (TACE), radiofrequency ablation, and radiotherapy, induce tumor cell necrosis to release tumor-associated antigens (TAAs), increase tumor immunogenicity, activate antitumor immune responses, and increase the number of tumor-infiltrating cytotoxic CD8+ T cells, thereby generating synergistic effects with ICIs[131]. Even for recurrent HCC, LRTs combined with ICIs improve patient prognosis by remodeling the TME and reducing tumor burden[132]. Furthermore, novel ablation techniques such as irreversible electroporation[133] and nanosecond pulsed electric fields[134] disrupt tumor cell membranes to enhance antigen presentation, further optimizing the efficacy of ICIs in treating HCC.
Drug combination therapy
Combinatorial strategies targeting TME remodeling through multiple mechanisms are pivotal in overcoming HCC resistance to ICIs.
The atezolizumab-bevacizumab regimen, established as a first-line therapy for advanced HCC, exerts efficacy via angiogenic normalization[135]. Bevacizumab rectifies aberrant vasculature to enhance CD8+/Th-1 cell infiltration, converting immunosuppressive to immunologically active TME states, as evidenced by the IMBrave150 trial demonstrating superior OS and PFS vs. sorafenib (objective response rate, ORR: 30%)[136]. The CARES-310 trial further validated the synergy of tyrosine kinase inhibitor (TKI)-ICI, with camrelizumab-apatinib achieving a median PFS of 5.6 vs. 3.7 months with sorafenib in unresectable/metastatic HCC (n = 543), alongside an enhanced ORR and disease control rate (DCR)[137]. Furthermore, dual PD-1/PD-L1 and TIGIT blockade augments intratumoral cytotoxic T lymphocyte infiltration through concurrent inhibition of T cell exhaustion pathways, effectively curtailing tumor progression[138].
Dual checkpoint inhibition strategies in HCC leverage synergistic targeting of complementary immune pathways. The nivolumab-ipilimumab (“O + Y”) regimen combines PD-1 blockade (alleviating PD-L1-mediated T cell suppression) with CTLA-4 inhibition (expanding T cell diversity through CTLA-4 blockade and Treg depletion), demonstrating clinical efficacy in the CheckMate-9DW trial, with a 36% ORR, 30.4-month median duration of response (DoR), and 38% 3-year OS with manageable toxicity[139]. This paradigm has established dual immunotherapy as a first-line HCC therapy in China.
The tremelimumab-durvalumab (“STRIDE”) regimen employs a phased approach: single high-dose CTLA-4 inhibition (tremelimumab) primes acute immune activation, whereas sustained PD-L1 blockade (durvalumab) maintains durable antitumor immunity with reduced toxicity compared with conventional dual inhibition[140]. TIM-3 co-targeting has also been explored. Preclinical studies have shown that TIM-3/PD-1 dual blockade in HCC mouse models significantly inhibits tumor progression, remodels the immune microenvironment, enhances T cell infiltration, and reduces PD-1/TIM-3 expression on CD8+ T cells, increasing ICI efficacy[141].
These combinatorial approaches share a unified rationale: multidimensional modulation of angiogenesis, T cell exhaustion, and immunosuppressive cell populations to reverse TME immunosuppression, opening new therapeutic windows for resistant patients. Future efforts should prioritize biomarker-guided personalized combinations and optimize treatment sequencing to balance efficacy and safety.
New strategies targeting drug resistance mechanisms
Emerging strategies to overcome ICI resistance in HCC focus on metabolic vulnerabilities, intrinsic resistance pathways, and extrinsic immunosuppressive networks. Metabolic targeting of TK1/LDHA and lactate accumulation reprograms immunosuppressive TMEs, with TK1 inhibitors synergizing with PD-1 blockade to enhance cytotoxic T cell infiltration and suppress tumor growth in preclinical models[29], while inhibition of LPCAT1 or CERS5 restores NK cell cytotoxicity[30]. Spatial immune mapping via TIMES scoring can be used to predict recurrence risk and guide personalized therapy[142]. Intrinsic resistance mechanisms include high-affinity neoantigen (HAN)-CD39+CD8+ TIL prognostic models that optimize PD-1 inhibitor stratification[34], FASN inhibition to prevent palmitoylation-mediated MHC-I lysosomal degradation[72], and IRGQ-targeted autophagy modulation to restore antigen presentation[67]. To address signaling dysregulation, a supramolecular polypeptide that targets both β-catenin and PD-L1 has been recently developed and has demonstrated potent antitumor efficacy and biosafety in HCC immunotherapy[143]. Targeting MerTK with inhibitors such as sitravatinib synergizes with ICIs to induce ferroptosis in PD-L1-resistant TMEs, reduce MDSC recruitment, and activate CD8+ T cells[81]. Notably, silencing BIRC2 via shRNA or small molecules sensitizes HCC cells to immune killing, improves T cell function, and enhances ICI efficacy in preclinical models[87].
With respect to extrinsic resistance, researchers have investigated several strategies. MDSC accumulation is strongly associated with ICI resistance[86]. CCRK overexpression in tumors promotes MDSC accumulation and is correlated with poor prognosis; CCRK inhibition reverses the immunosuppressive TME[144]. As CRKL contributes to the immunosuppressive TME, CRKL knockout or combined bevacizumab therapy restores anti-PD-1 efficacy in HCC mouse models, supporting the use of CRKL inhibitors plus ICIs as a promising approach[91]. ACE2 modulates the TME by suppressing M2-like macrophage polarization and sensitizing tumors to anti-PD-L1 therapy, highlighting the ACE2 axis as a potential immunomodulatory target[92]. Norathyriol, a metabolic inhibitor, reverses circPETH-147aa-driven metabolic and metastatic phenotypes, enhances CD8+ T cell cytotoxicity, and synergizes with ICIs to counteract metabolic immune evasion in advanced HCC[93]. In addition to TIM-3/PD-1 dual blockade, a peptide inhibitor targeting TIM-3 palmitoylation enhances CAR-T and NK cell antitumor activity in preclinical models, suggesting a novel therapeutic avenue for HCC[96].
CONCLUSION
Despite the demonstrated clinical benefits of ICIs in HCC, therapeutic resistance remains a major barrier to durable efficacy. This review delineates ICI applications in HCC through a tripartite framework: resistance mechanisms (intrinsic/extrinsic pathways, immunosuppressive microenvironment remodeling), LT challenges (graft rejection, immunosuppressive regimen optimization), and strategic countermeasures (biomarker-guided combinations, novel target development). Future priorities include elucidating resistance evolution via multiomics profiling, designing rational therapeutic combinations, and advancing the clinical translation of emerging modalities. Crucially, bridging mechanistic insights from preclinical models with biomarker-adaptive clinical trials will enable precision immunotherapy to balance antitumor efficacy and transplant-related immune tolerance, ultimately improving survival outcomes through vertically integrated basic–clinical research paradigms.
DECLARATIONS
Acknowledgments
The authors thank BioGDP. The Graphic Abstract and figures were created with BioGDP.com.
Authors’ contributions
Conceptualization: Li H, Ye L, Chen Y
Writing - original draft preparation: Chen Y
Writing - review and editing: Chen Y, Chen D, Liang Z, Yu H, Sun H, Hu Y, Jiang P, Zhang M
Funding acquisition and supervision: Li H, Ye L
All authors read and approved the final manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This research was supported by the Guangzhou Key Research and Development Program of the Guangzhou Municipal Science and Technology Bureau (Grant no. 02A004999000186).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16:589-604.
2. Kohansal-Nodehi M, Swiatek-de Lange M, Kroeniger K, et al. Discovery of a haptoglobin glycopeptides biomarker panel for early diagnosis of hepatocellular carcinoma. Front Oncol. 2023;13:1213898.
3. Zheng Y, Wang S, Cai J, Ke A, Fan J. The progress of immune checkpoint therapy in primary liver cancer. Biochim Biophys Acta Rev Cancer. 2021;1876:188638.
4. Llovet JM, Castet F, Heikenwalder M, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19:151-72.
5. Akula V, Chen L, Acikgoz Y, et al. Neoadjuvant immune checkpoint inhibitors for hepatocellular carcinoma. NPJ Precis Oncol. 2025;9:60.
6. Shek D, Read SA, Nagrial A, et al. Immune-checkpoint inhibitors for advanced hepatocellular carcinoma: a synopsis of response rates. Oncologist. 2021;26:e1216-25.
7. De Lorenzo S, Tovoli F, Trevisani F. Mechanisms of primary and acquired resistance to immune checkpoint inhibitors in patients with hepatocellular carcinoma. Cancers. 2022;14:4616.
8. Zheng Y, Wang T, Tu X, et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J Immunother Cancer. 2019;7:193.
9. Ortiz V, Loeuillard E. Rethinking immune check point inhibitors use in liver transplantation: implications and resistance. Cell Mol Gastroenterol Hepatol. 2025;19:101407.
10. Yin C, Baba T, He AR, Smith C. Immune checkpoint inhibitors in liver transplant recipients - a review of current literature. Hepatoma Res. 2021;7:52.
11. Oura K, Morishita A, Tani J, Masaki T. Tumor immune microenvironment and immunosuppressive therapy in hepatocellular carcinoma: a review. Int J Mol Sci. 2021;22:5801.
12. Shen KY, Zhu Y, Xie SZ, Qin LX. Immunosuppressive tumor microenvironment and immunotherapy of hepatocellular carcinoma: current status and prospectives. J Hematol Oncol. 2024;17:25.
13. Ping Y, Shan J, Qin H, et al. PD-1 signaling limits expression of phospholipid phosphatase 1 and promotes intratumoral CD8+ T cell ferroptosis. Immunity. 2024;57:2122-39.e9.
14. Pu Q, Yu L, Liu X, et al. Prognostic value of CD8+ T cells related genes and exhaustion regulation of Notch signaling pathway in hepatocellular carcinoma. Front Immunol. 2024;15:1375864.
15. Song G, Shi Y, Zhang M, et al. Global immune characterization of HBV/HCV-related hepatocellular carcinoma identifies macrophage and T-cell subsets associated with disease progression. Cell Discov. 2020;6:90.
16. Guo H, Wang M, Ni C, et al. TREM2 promotes the formation of a tumor-supportive microenvironment in hepatocellular carcinoma. J Exp Clin Cancer Res. 2025;44:20.
17. Peña-Romero AC, Orenes-Piñero E. Dual effect of immune cells within tumour microenvironment: pro- and anti-tumour effects and their triggers. Cancers. 2022;14:1681.
18. Mempel TR, Lill JK, Altenburger LM. How chemokines organize the tumour microenvironment. Nat Rev Cancer. 2024;24:28-50.
20. Langhans B, Nischalke HD, Krämer B, et al. Role of regulatory T cells and checkpoint inhibition in hepatocellular carcinoma. Cancer Immunol Immunother. 2019;68:2055-66.
21. Nan J, Xing YF, Hu B, et al. Endoplasmic reticulum stress induced LOX-1+ CD15+ polymorphonuclear myeloid-derived suppressor cells in hepatocellular carcinoma. Immunology. 2018;154:144-55.
22. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer. 2020;19:116.
23. Wei R, Song J, Liu C, et al. FAP upregulates PD-L1 expression in cancer-associated fibroblasts to exacerbate T cells dysfunction and suppress anti-tumor immunity. Cancer Lett. 2025;612:217475.
24. Raimondo S, Pucci M, Alessandro R, Fontana S. Extracellular vesicles and tumor-immune escape: biological functions and clinical perspectives. Int J Mol Sci. 2020;21:2286.
25. Vautrot V, Bentayeb H, Causse S, Garrido C, Gobbo J. Tumor-derived exosomes: hidden players in PD-1/PD-L1 resistance. Cancers. 2021;13:4537.
26. Sun Y, Wu L, Zhong Y, et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell. 2021;184:404-21.e16.
27. Zheng C, Zheng L, Yoo JK, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169:1342-56.e16.
28. Lin J, Rao D, Zhang M, Gao Q. Metabolic reprogramming in the tumor microenvironment of liver cancer. J Hematol Oncol. 2024;17:6.
29. Li Q, Zhang L, Yang Q, et al. Thymidine kinase 1 drives hepatocellular carcinoma in enzyme-dependent and -independent manners. Cell Metab. 2023;35:912-27.e7.
30. Liu Q, Zhang X, Qi J, et al. Comprehensive profiling of lipid metabolic reprogramming expands precision medicine for HCC. Hepatology. 2025;81:1164-80.
31. Galassi C, Chan TA, Vitale I, Galluzzi L. The hallmarks of cancer immune evasion. Cancer Cell. 2024;42:1825-63.
32. Jung J, Heo YJ, Park S. High tumor mutational burden predicts favorable response to anti-PD-(L)1 therapy in patients with solid tumor: a real-world pan-tumor analysis. J Immunother Cancer. 2023;11:e006454.
33. Anagnostou V, Bardelli A, Chan TA, Turajlic S. The status of tumor mutational burden and immunotherapy. Nat Cancer. 2022;3:652-6.
34. Chan TA, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30:44-56.
35. Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202-6.
37. Li L, Wang B, Zhao S, Xiong Q, Cheng A. The role of ANXA1 in the tumor microenvironment. Int Immunopharmacol. 2024;131:111854.
38. Liu P, Zhao L, Loos F, et al. Immunosuppression by mutated calreticulin released from malignant cells. Mol Cell. 2020;77:748-60.e9.
39. Galluzzi L, Guilbaud E, Schmidt D, Kroemer G, Marincola FM. Targeting immunogenic cell stress and death for cancer therapy. Nat Rev Drug Discov. 2024;23:445-60.
40. Yang W, Feng Y, Zhou J, et al. A selective HDAC8 inhibitor potentiates antitumor immunity and efficacy of immune checkpoint blockade in hepatocellular carcinoma. Sci Transl Med. 2021;13:eaaz6804.
41. Teijeira Á, Garasa S, Gato M, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52:856-71.e8.
42. Peranzoni E, Lemoine J, Vimeux L, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A. 2018;115:E4041-50.
43. Ho P, Melms JC, Rogava M, et al. The CD58-CD2 axis is co-regulated with PD-L1 via CMTM6 and shapes anti-tumor immunity. Cancer Cell. 2023;41:1207-21.e12.
44. Chan LC, Li CW, Xia W, et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J Clin Invest. 2019;129:3324-38.
45. Liu X, Song J, Zhang H, et al. Immune checkpoint HLA-E:CD94-NKG2A mediates evasion of circulating tumor cells from NK cell surveillance. Cancer Cell. 2023;41:272-87.e9.
46. Ritchie C, Carozza JA, Li L. Biochemistry, cell biology, and pathophysiology of the innate immune cGAS-cGAMP-STING pathway. Annu Rev Biochem. 2022;91:599-628.
47. Hung MH, Lee JS, Ma C, et al. Tumor methionine metabolism drives T-cell exhaustion in hepatocellular carcinoma. Nat Commun. 2021;12:1455.
48. Ockfen E, Filali L, Pereira Fernandes D, Hoffmann C, Thomas C. Actin cytoskeleton remodeling at the cancer cell side of the immunological synapse: good, bad, or both? Front Immunol. 2023;14:1276602.
49. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021;6:402.
50. Wang H, Sun P, Yuan X, et al. Autophagy in tumor immune escape and immunotherapy. Mol Cancer. 2025;24:85.
51. Lim CJ, Lee YH, Pan L, et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut. 2019;68:916-27.
52. Gao Y, Zhang Z, Huang X, et al. HBV-associated hepatocellular carcinomas inhibit antitumor CD8+ T cell via the long noncoding RNA HDAC2-AS2. Nat Commun. 2025;16:2055.
53. Yang C, Geng H, Yang X, et al. Targeting the immune privilege of tumor-initiating cells to enhance cancer immunotherapy. Cancer Cell. 2024;42:2064-81.e19.
54. Wang G, Shen X, Jin W, et al. Elucidating the role of S100A10 in CD8+ T cell exhaustion and HCC immune escape via the cPLA2 and 5-LOX axis. Cell Death Dis. 2024;15:573.
55. Zhang Z, Huang W, Hu D, et al. E-twenty-six-specific sequence variant 5 (ETV5) facilitates hepatocellular carcinoma progression and metastasis through enhancing polymorphonuclear myeloid-derived suppressor cell (PMN-MDSC)-mediated immunosuppression. Gut. 2025;74:1137-49.
56. Zhang Y, Wang M, Ye L, et al. HKDC1 promotes tumor immune evasion in hepatocellular carcinoma by coupling cytoskeleton to STAT1 activation and PD-L1 expression. Nat Commun. 2024;15:1314.
57. Jiang X, Deng W, Tao S, et al. A RIPK3-independent role of MLKL in suppressing parthanatos promotes immune evasion in hepatocellular carcinoma. Cell Discov. 2023;9:7.
58. Nguyen PHD, Wasser M, Tan CT, et al. Trajectory of immune evasion and cancer progression in hepatocellular carcinoma. Nat Commun. 2022;13:1441.
59. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci U S A. 2021;118:e2023739118.
60. Graydon CG, Mohideen S, Fowke KR. LAG3’s enigmatic mechanism of action. Front Immunol. 2020;11:615317.
61. Shang S, Zhao Y, Qian K, et al. The role of neoantigens in tumor immunotherapy. Biomed Pharmacother. 2022;151:113118.
62. Klempner SJ, Fabrizio D, Bane S, et al. Tumor mutational burden as a predictive biomarker for response to immune checkpoint inhibitors: a review of current evidence. Oncologist. 2020;25:e147-59.
63. Ang C, Klempner SJ, Ali SM, et al. Prevalence of established and emerging biomarkers of immune checkpoint inhibitor response in advanced hepatocellular carcinoma. Oncotarget. 2019;10:4018-25.
64. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707-23.
65. Xu J, Zhang Y, Jia R, et al. Anti-PD-1 antibody SHR-1210 combined with apatinib for advanced hepatocellular carcinoma, gastric, or esophagogastric junction cancer: an open-label, dose escalation and expansion study. Clin Cancer Res. 2019;25:515-23.
66. Zhu AX, Abbas AR, de Galarreta MR, et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat Med. 2022;28:1599-611.
67. Herhaus L, Gestal-Mato U, Eapen VV, et al. IRGQ-mediated autophagy in MHC class I quality control promotes tumor immune evasion. Cell. 2024;187:7285-302.e29.
68. Akazawa Y, Nobuoka D, Takahashi M, et al. Higher human lymphocyte antigen class I expression in early-stage cancer cells leads to high sensitivity for cytotoxic T lymphocytes. Cancer Sci. 2019;110:1842-52.
69. Umemoto Y, Okano S, Matsumoto Y, et al. Prognostic impact of programmed cell death 1 ligand 1 expression in human leukocyte antigen class I-positive hepatocellular carcinoma after curative hepatectomy. J Gastroenterol. 2015;50:65-75.
70. Yoshida S, Hazama S, Tokuno K, et al. Concomitant overexpression of heat-shock protein 70 and HLA class-I in hepatitis C virus-related hepatocellular carcinoma. Anticancer Res. 2009;29:539-44.
71. Yao H, Lan J, Li C, et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat Biomed Eng. 2019;3:306-17.
72. Huang J, Tsang WY, Fang XN, et al. FASN inhibition decreases MHC-I degradation and synergizes with PD-L1 checkpoint blockade in hepatocellular carcinoma. Cancer Res. 2024;84:855-71.
73. Ruiz de Galarreta M, Bresnahan E, Molina-Sánchez P, et al. β-Catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019;9:1124-41.
74. Li X, Xiang Y, Li F, Yin C, Li B, Ke X. WNT/β-Catenin signaling pathway regulating T cell-inflammation in the tumor microenvironment. Front Immunol. 2019;10:2293.
75. Takeuchi Y, Tanegashima T, Sato E, et al. Highly immunogenic cancer cells require activation of the WNT pathway for immunological escape. Sci Immunol. 2021;6:eabc6424.
76. Cadoux M, Caruso S, Pham S, et al. Expression of NKG2D ligands is downregulated by β-catenin signalling and associates with HCC aggressiveness. J Hepatol. 2021;74:1386-97.
77. Harding JJ, Nandakumar S, Armenia J, et al. Prospective genotyping of hepatocellular carcinoma: clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin Cancer Res. 2019;25:2116-26.
78. Pinyol R, Sia D, Llovet JM. Immune exclusion-Wnt/CTNNB1 class predicts resistance to immunotherapies in HCC. Clin Cancer Res. 2019;25:2021-3.
79. Shin DS, Zaretsky JM, Escuin-Ordinas H, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7:188-201.
80. Tsimberidou AM, Vining DJ, Arora SP, et al. Phase I trial of TTI-101, a first-in-class oral inhibitor of STAT3, in patients with advanced solid tumors. Clin Cancer Res. 2025;31:965-74.
81. Wang S, Zhu L, Li T, et al. Disruption of MerTK increases the efficacy of checkpoint inhibitor by enhancing ferroptosis and immune response in hepatocellular carcinoma. Cell Rep Med. 2024;5:101415.
82. Xu Y, Poggio M, Jin HY, et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat Med. 2019;25:301-11.
83. Montironi C, Castet F, Haber PK, et al. Inflamed and non-inflamed classes of HCC: a revised immunogenomic classification. Gut. 2023;72:129-40.
84. Shen J, Ju Z, Zhao W, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. 2018;24:556-62.
85. Li J, Wang W, Zhang Y, et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J Clin Invest. 2020;130:2712-26.
86. Zhou J, Liu M, Sun H, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut. 2018;67:931-44.
87. Fu L, Li S, Mei J, et al. BIRC2 blockade facilitates immunotherapy of hepatocellular carcinoma. Mol Cancer. 2025;24:113.
88. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74-80.
89. Tian X, Shen H, Li Z, Wang T, Wang S. Tumor-derived exosomes, myeloid-derived suppressor cells, and tumor microenvironment. J Hematol Oncol. 2019;12:84.
90. Hoechst B, Voigtlaender T, Ormandy L, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50:799-807.
91. Xie P, Yu M, Zhang B, et al. CRKL dictates anti-PD-1 resistance by mediating tumor-associated neutrophil infiltration in hepatocellular carcinoma. J Hepatol. 2024;81:93-107.
92. Xie P, Guo L, Yu Q, et al. ACE2 enhances sensitivity to PD-L1 blockade by inhibiting macrophage-induced immunosuppression and angiogenesis. Cancer Res. 2025;85:299-313.
93. Lan T, Gao F, Cai Y, et al. The protein circPETH-147aa regulates metabolic reprogramming in hepatocellular carcinoma cells to remodel immunosuppressive microenvironment. Nat Commun. 2025;16:333.
94. Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol. 2016;27:1492-504.
95. Bai X, Yi M, Jiao Y, Chu Q, Wu K. Blocking TGF-β signaling to enhance the efficacy of immune checkpoint inhibitor. Onco Targets Ther. 2019;12:9527-38.
96. Zhang Z, Ren C, Xiao R, et al. Palmitoylation of TIM-3 promotes immune exhaustion and restrains antitumor immunity. Sci Immunol. 2024;9:eadp7302.
97. Ganjalikhani Hakemi M, Jafarinia M, Azizi M, Rezaeepoor M, Isayev O, Bazhin AV. The role of TIM-3 in hepatocellular carcinoma: a promising target for immunotherapy? Front Oncol. 2020;10:601661.
98. Jiang Y, Dai A, Huang Y, et al. Ligand-induced ubiquitination unleashes LAG3 immune checkpoint function by hindering membrane sequestration of signaling motifs. Cell. 2025;188:2354-71.e18.
99. Terry S, Savagner P, Ortiz-Cuaran S, et al. New insights into the role of EMT in tumor immune escape. Mol Oncol. 2017;11:824-46.
100. Soundararajan R, Fradette JJ, Konen JM, et al. Targeting the interplay between epithelial-to-mesenchymal-transition and the immune system for effective immunotherapy. Cancers. 2019;11:714.
101. Pallotta MT, Rossini S, Suvieri C, et al. Indoleamine 2,3-dioxygenase 1 (IDO1): an up-to-date overview of an eclectic immunoregulatory enzyme. FEBS J. 2022;289:6099-118.
102. Xue HY, Wei F. TGF-β: an active participant in the immune and metabolic microenvironment of multiple myeloma: TGF-β in the microenvironment of multiple myeloma. Ann Hematol. 2024;103:4351-62.
103. Cherney EC, Zhang L, Nara S, et al. Discovery and preclinical evaluation of BMS-986242, a potent, selective inhibitor of indoleamine-2,3-dioxygenase 1. ACS Med Chem Lett. 2021;12:288-94.
104. Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. 2020;37:443-55.
105. Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819-29.
106. Sade-Feldman M, Jiao YJ, Chen JH, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8:1136.
107. Gettinger S, Choi J, Hastings K, et al. Impaired HLA Class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017;7:1420-35.
108. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409-13.
109. Trujillo JA, Luke JJ, Zha Y, et al. Secondary resistance to immunotherapy associated with β-catenin pathway activation or PTEN loss in metastatic melanoma. J Immunother Cancer. 2019;7:295.
110. Zhao F, Xiao C, Evans KS, et al. Paracrine Wnt5a-β-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity. 2018;48:147-60.e7.
111. Kakavand H, Jackett LA, Menzies AM, et al. Negative immune checkpoint regulation by VISTA: a mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod Pathol. 2017;30:1666-76.
112. Wang B, Han Y, Zhang Y, et al. Overcoming acquired resistance to cancer immune checkpoint therapy: potential strategies based on molecular mechanisms. Cell Biosci. 2023;13:120.
113. Spranger S. Tumor heterogeneity and tumor immunity: a chicken-and-egg problem. Trends Immunol. 2016;37:349-51.
114. Ling YH, Liu MN, Yin YX, et al. Integrated genetic and epigenetic analysis reveals DNA repair alterations in multifocal hepatocellular carcinoma. Signal Transduct Target Ther. 2023;8:244.
115. Ma C, Han M, Heinrich B, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360:eaan5931.
116. Yoshimoto S, Loo TM, Atarashi K, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97-101.
117. Vétizou M, Pitt JM, Daillère R, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079-84.
118. Bianca C, Sidhartha E, Tiribelli C, El-Khobar KE, Sukowati CHC. Role of hepatitis B virus in development of hepatocellular carcinoma: focus on covalently closed circular DNA. World J Hepatol. 2022;14:866-84.
119. Kao C, Charmsaz S, Tsai HL, et al. Age-related divergence of circulating immune responses in patients with solid tumors treated with immune checkpoint inhibitors. Nat Commun. 2025;16:3531.
120. Ho JK, Thurairajah PH, Leo J, Huang DQ, Fan KH. Sex differences in hepatocellular carcinoma. Hepatoma Res. 2024;10:53.
121. Qiao ZY, Zhang ZJ, Lv ZC, et al. Neoadjuvant programmed cell death 1 (PD-1) inhibitor treatment in patients with hepatocellular carcinoma before liver transplant: a cohort study and literature review. Front Immunol. 2021;12:653437.
122. Moeckli B, Wassmer CH, El Hajji S, et al. Determining safe washout period for immune checkpoint inhibitors prior to liver transplantation: an international retrospective cohort study. Hepatology. 2025.
123. Kayali S, Pasta A, Plaz Torres MC, et al. Immune checkpoint inhibitors in malignancies after liver transplantation: a systematic review and pooled analysis. Liver Int. 2023;43:8-17.
124. Au KP, Chok KSH. Immunotherapy after liver transplantation: where are we now? World J Gastrointest Surg. 2021;13:1267-78.
125. Guo Z, Liu Y, Ling Q, et al. Pretransplant use of immune checkpoint inhibitors for hepatocellular carcinoma: a multicenter, retrospective cohort study. Am J Transplant. 2024;24:1837-56.
126. Abdelrahim M, Esmail A, Divatia MK, et al. Utilization of immunotherapy as a neoadjuvant therapy for liver transplant recipients with hepatocellular carcinoma. J Clin Med. 2024;13:3068.
127. Jin X, Ma X, Zhao D, Yang L, Ma N. Immune microenvironment and therapeutic progress of recurrent hepatocellular carcinoma after liver transplantation. Transl Oncol. 2023;28:101603.
128. Li J, Zhang L, Xing H, et al. The absence of intra-tumoral tertiary lymphoid structures is associated with a worse prognosis and mTOR signaling activation in hepatocellular carcinoma with liver transplantation: a multicenter retrospective study. Adv Sci. 2024;11:e2309348.
129. Martínez Burgos M, González Grande R, López Ortega S, et al. Liver transplantation for hepatocarcinoma: results over two decades of a transplantation programme and analysis of factors associated with recurrence. Biomedicines. 2024;12:1302.
130. Shi GM, Wang J, Huang XW, et al. Graft programmed death ligand 1 expression as a marker for transplant rejection following anti-programmed death 1 immunotherapy for recurrent liver tumors. Liver Transpl. 2021;27:444-9.
131. Dendy MS, Ludwig JM, Stein SM, Kim HS. Locoregional therapy, immunotherapy and the combination in hepatocellular carcinoma: future directions. Liver Cancer. 2019;8:326-40.
132. Liang J, Bai Y, Ha FS, Luo Y, Deng HT, Gao YT. Combining local regional therapy and systemic therapy: expected changes in the treatment landscape of recurrent hepatocellular carcinoma. World J Gastrointest Oncol. 2023;15:1-18.
133. Dai Z, Wang Z, Lei K, et al. Irreversible electroporation induces CD8+ T cell immune response against post-ablation hepatocellular carcinoma growth. Cancer Lett. 2021;503:1-10.
134. Qian J, Chen T, Wu Q, et al. Blocking exposed PD-L1 elicited by nanosecond pulsed electric field reverses dysfunction of CD8+ T cells in liver cancer. Cancer Lett. 2020;495:1-11.
135. Cheng AL, Qin S, Ikeda M, et al. Updated efficacy and safety data from IMbrave150: atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol. 2022;76:862-73.
136. Galle PR, Finn RS, Qin S, et al. Patient-reported outcomes with atezolizumab plus bevacizumab versus sorafenib in patients with unresectable hepatocellular carcinoma (IMbrave150): an open-label, randomised, phase 3 trial. Lancet Oncol. 2021;22:991-1001.
137. Qin S, Chan SL, Gu S, et al; CARES-310 Study Group. Camrelizumab plus rivoceranib versus sorafenib as first-line therapy for unresectable hepatocellular carcinoma (CARES-310): a randomised, open-label, international phase 3 study. Lancet. 2023;402:1133-46.
138. Li Y, Li B, Wang Q, et al. Dual targeting of TIGIT and PD-1 with a novel small molecule for cancer immunotherapy. Biochem Pharmacol. 2024;223:116162.
139. Yau T, Galle PR, Decaens T, et al; CheckMate 9DW investigators. Nivolumab plus ipilimumab versus lenvatinib or sorafenib as first-line treatment for unresectable hepatocellular carcinoma (CheckMate 9DW): an open-label, randomised, phase 3 trial. Lancet. 2025;405:1851-64.
140. Abou-Alfa GK, Lau G, Kudo M, et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. NEJM Evid. 2022;1:EVIDoa2100070.
141. Liu F, Liu Y, Chen Z. Tim-3 expression and its role in hepatocellular carcinoma. J Hematol Oncol. 2018;11:126.
142. Jia G, He P, Dai T, et al. Spatial immune scoring system predicts hepatocellular carcinoma recurrence. Nature. 2025;640:1031-41.
143. Zhou Z, Li X, Yang G, et al. Targeting β-catenin and PD-L1 simultaneously by a racemic supramolecular peptide for the potent immunotherapy of hepatocellular carcinoma. Theranostics. 2023;13:3371-86.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.