


Exploring topological materials for hydrogen evolution reaction: insights from density functional theory
 0
0 Abstract
Hydrogen energy technologies offer a transformative shift toward reducing reliance on fossil fuels and creating a sustainable, low-carbon future. In this shift, topological materials, known for their strong electron interactions and unique physical properties, present promising opportunities in electrocatalysis. In this study, we performed a systematic density functional theory analysis of over 100 topological materials and examined more than 1,000 adsorption sites. Our findings reveal that topological materials possess abundant and diverse active sites, resulting in a wide range of hydrogen adsorption energies ranging from -1.5 eV to 0 eV. To identify the most promising catalysts for hydrogen evolution reaction (HER) in acidic media, we focused on the topological materials with hydrogen adsorption energies within -0.27 ± 0.1 eV. The Gibbs free energy of hydrogen adsorption (ΔGH*) was evaluated for the HER. All selected materials showed ΔGH* values between -0.31 and -0.16 eV. Based on these results, 11 promising candidates were identified with high potential for efficient HER activity. Our study establishes fundamental structure-property-activity relationships that can serve as a reliable dataset for further machine-learning studies, while also providing valuable insights and design guidelines for the continued exploration of topological materials as high-performance HER catalysts.
Keywords
INTRODUCTION
Hydrogen energy technologies have been recognized as a promising candidate to reduce dependence on fossil fuels and meet future energy demands[1,2]. Currently, fossil fuels, including natural gas, coal, and oil, account for approximately 80% of the world’s energy consumption[3]. The combustion of fossil fuels releases large amounts of greenhouse gases, leading to more severe weather events, rising sea levels, and declining biodiversity. Additionally, it also fuels geopolitical conflicts and energy security concerns due to the unequal distribution of reserves[4,5]. In response to these growing environmental concerns and energy security challenges, hydrogen has the potential to drive the global transition toward a more sustainable, low-carbon future[6].
The development of highly efficient, selective, stable, and cost-effective catalysts for the water-splitting process is the crucial step toward a thriving hydrogen economy. Utilizing electrocatalysts along with electricity from renewable sources for the hydrogen evolution reaction (HER) is one of the most promising approaches. Platinum-group metals, especially platinum and its alloys, have emerged as the most effective electrocatalysts for this process[7-9]. Among them, the ultralow-Pt-loading electrocatalysts, which can reach the balance of low cost and high HER performance, have attracted much attention. Chen et al. demonstrated that anchoring platinum clusters onto two-dimensional fullerene nanosheets (PtC60) significantly boosts platinum’s activity for the alkaline HER. This PtC60 composite shows 12 times higher intrinsic activity compared to the traditional Pt/C catalyst in alkaline conditions[10]. Smiljanić et al. reported a novel composite catalyst consisting of Pt nanoparticles supported on TiONx (Pt/TiONx), which outperformed the benchmark Pt/C catalyst regarding both HER activity and stability in acid electrolytes[11]. Other transition metals (Co, Fe, Mo, Ni, etc.) have also been explored as effective electrocatalysts for HER in recent years to confront the scarcity and high cost of Pt[12-15]. At the same time, single-atom catalysts (SACs) are receiving increasing attention due to the maximized atom utilization efficiency arising from their abundant active sites[16-18]. Yet, the search for electrocatalysts that simultaneously combine high activity, selectivity, stability, and cost-effectiveness remains ongoing.
Topological materials, known for their strong electron interactions and distinctive physical properties, provide exciting new opportunities in the field of electrocatalysis[19,20]. With their unique surface states and exceptional charge mobility, they emerge as highly effective electrocatalysts capable of addressing the limitations of current hydrogen production technologies[21]. Qu et al. found that Bi2Te3 topological insulator thin films, when having partially oxidized surfaces or tellurium vacancies, show high activity for HER, with a current density reaching up to 1.74 μA cm-2 for a 48 nm Bi2Te3 thin film[22]. Through first-principles calculations, Li et al. reported a new topological nodal line semimetal, namely the TiSi-type family, which exhibits a closed Dirac nodal line arising from linear band crossings in the ky = 0 plane. The hydrogen adsorbed state on the corresponding surface yields ΔGH* (ΔGH* represents the Gibbs free energy of hydrogen adsorption, where * denotes an active site on the catalyst surface) close to zero, indicating near-optimal activity for HER[23]. Our group has explored the topological nodal-line semimetal AuSn4, discovering that oxidized AuSn4 can be an effective catalyst for HER in alkaline media[24].
Despite numerous efforts, the search for the optimal electrocatalyst for HER remains ongoing; the structure-property-activity relationships of topological materials are still missing. In this work, we conduct density functional theory (DFT) calculations to systematically screen topological materials, aiming to identify high-performance topological material catalysts specifically suited for HER. Our study screens 1,000 sites across nearly 100 surfaces of topological materials and identifies 11 topological materials promising as superior electrocatalysts for HER. Our study not only helps better understand topological materials and their relationship to HER but also provides guidance for future research in material synthesis and electrochemistry toward developing more effective HER catalysts.
CALCULATION METHOD
The DFT calculations were carried out by using the plane-wave-based Vienna Ab initio Simulation Package (VASP 5.4.1)[25,26]. The exchange-correlation interactions were modeled using the Perdew-Burke-Ernzerhof (PBE) functional within the framework of the generalized gradient approximation (GGA)[27]. This functional has been widely demonstrated to provide a reliable description of the electronic structure and energetics of solid-state and surface systems. The interactions between core and electrons were described with the projector augmented wave (PAW) method[28-30], enabling an accurate yet computationally efficient representation of the ionic cores. All atomic structures were fully optimized prior to property calculations, with both atomic positions allowed to relax until the residual forces on each atom were smaller than
RESULTS AND DISCUSSION
Adsorption sites
The intricate surface structure of topological materials provides rich reaction sites, making these materials promising candidates in the field of catalysis. Given that low-index crystal facets are more energetically favorable and thus more amenable to synthesis, this study focuses on all the low-index crystal facets, including (001), (100), (101), (110), and (111) surfaces [Figure 1]. These facets differ markedly in their atomic arrangements, surface atom coordination numbers, and bonding environments. Such variations lead to different local electronic structures and adsorption geometries, thereby creating diverse reaction conditions across the surface, which may result in more active sites available for enhanced activities.
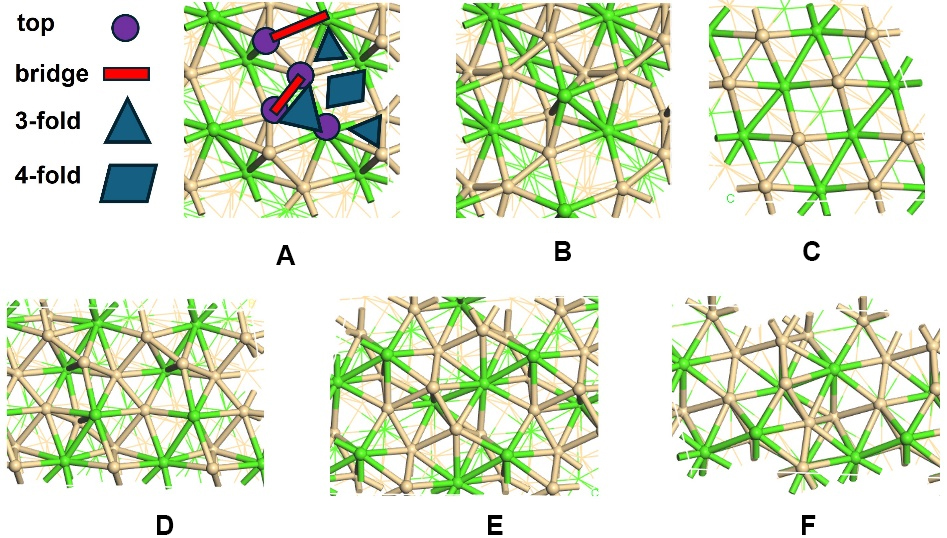
Figure 1. All the possible low-index facets and adsorption sites studied in this work. (A and B) Cr1Ce1 (001) surface with Cr and Ce termination, respectively; (C-F): (100), (101), (110) and (111) surfaces. Green and ochre spheres represent Cr and Ce atoms, respectively. The different adsorption sites: top, bridge, 3-fold and 4-fold hollow sites are illustrated in Figure 1A.
Distinct surface terminations give rise to a variety of adsorption sites, each of which can have a significant impact on HER performance. Therefore, it is essential to examine all possible adsorption sites to gain a complete and accurate understanding of the catalytic performance. To ensure a systematic and thorough study, our investigation carefully examines all potential reaction sites on each surface, encompassing metal, non-metal, and mixed terminations. For every termination, we consider all possible adsorption positions, including top sites located directly above individual surface atoms, bridge sites between two adjacent atoms, and hollow sites at the center of three or more neighboring atoms [Figure 1A]. A comprehensive analysis of catalytic performance at all the possible sites was conducted to identify the most stable adsorption site.
Adsorption energies
Adsorption plays a crucial role in surface reactions, as an ideal catalyst should have an intermediate binding affinity for reactants. The adsorption needs to be strong enough to facilitate the reaction but not so strong that it prevents the easy desorption of the products[33]. The adsorption energy of hydrogen on topological material varies in a wide range, as summarized in Table 1. As expected, the topological materials give abundant adsorption sites, leading to a wide range of adsorption energies for hydrogen. The most favorable adsorption sites also vary among different materials. On Al1Pt1 (001), the hydrogen prefers the Pt-top site, with a hydrogen adsorption energy (Eads) calculated to be 0.57 eV, indicating that hydrogen adsorption on Al1Pt1 is endothermic and not energetically favorable. As a result, Al1Pt1 (001) is expected to be inactive for HER because hydrogen adsorption is not energetically favorable, making subsequent reactions difficult. In contrast, on the Al1Au1 (110) surface, the hydrogen prefers the Al-top side, with an Eads calculated to be
Hydrogen adsorption energy (Eads, in eV) at the most favorable adsorption site for each material and surface termination; top, bridge, and hollow sites denoted by _t, _b, and _h, respectively
| Material | Eads | Sites | Material | Eads | Sites | Material | Eads | Sites |
| Al1Pd1_001 | -0.18 | Al-Pd-bri | Al1La1_111 | -0.88 | Al-La_b | C1Hf1_111 | -2.16 | Hf-Hf_h |
| Al1Pt1_001 | 0.57 | Pt-top | Ba1Sn1_111 | -0.38 | BaBaBa _h | C1Ti2_101 | -1.21 | Ti3_h |
| Au1Be1_001 | -0.07 | Au-Be_b | Ba2Zn1_111 | -0.47 | ZnZnZn_h | C1Ti2_110 | -1.19 | Ti-Ti_b |
| Co1Ge1_001 | -0.18 | Co_t | Bi1Li1_111 | -0.14 | Bi-Li_b | C1Ti2_111 | -1.27 | Ti3_h |
| Co1Si1_001 | -0.48 | Co-Co_b | Al1Au1_110 | -2.22 | Al_t | C1V2_101 | -1.05 | V-V_b |
| Cr1Ge1_001 | -0.42 | Cr-Cr_b | Al1La1_110 | -0.61 | Al-La_b | Cu1Ti2_001 | -1.06 | Cu3_h |
| Cr1Si1_001 | -0.39 | Cr-Cr_b | Al1Sc1_110 | -0.75 | Al-Al-Sc_h | Cu1Ti2_111 | -0.94 | CuTi_h |
| Fe1Pd1_001 | -0.94 | Fe_t | Al1La1_001 | -0.50 | La-La_b | Fe1Pt1_100 | -0.49 | Pt_t |
| Ga1Pt1_001 | -0.29 | Pt_t | Al1Pt2_001 | -0.78 | Al_t | Fe1Pt1_101 | -0.52 | Fe-Fe-Pt_h |
| Ge1Mn1_001 | -0.13 | Mn-Mn_b | Al1Sc1_0012 | -0.51 | AlAl_b | Fe1Pt1_110 | -4.20 | NA |
| Ge1Rh1_001 | -0.03 | Rh_t | As1Rh1_001 | -0.15 | Rh-Rh_b | Fe1Pt1_111 | -2.45 | Fe_t |
| Hf1Sb1_001 | -0.54 | Hf-Hf_b | As2U1_001 | -0.31 | As_t | Ga1Mn1_001 | -0.84 | Ga-Mn_b |
| Hf1Sn1_001 | -0.66 | Hf-Hf_b | C1Ni1_001 | -0.01 | Ni-Ni_h | Ga1Sc1_001 | -0.55 | Ga_t |
| Mn1Si1_001 | -0.26 | Mn-Si_b | C1W1_001 | -0.71 | W-W_h | Ga1Sc1_100 | -0.41 | Ga-Sc_b |
| Ni1Si1_001 | -0.20 | Si_t | C1Zr1_001 | -1.34 | Zr-Zr_h | Ga1Sc1_101 | -0.95 | Ga-Sc_b |
| Re1Si1_001 | -0.54 | Re_t | Ce1O1_001 | -1.02 | Ce-Ce_h | Ga1Sc1_110 | -1.17 | Ga-Sc_b |
| Si1V1_001 | -0.52 | V-V_b | H1V1_001 | -1.34 | V-V_h | Ga1Sc1_111 | -0.72 | Sc_t |
| Ga2Sc1_100 | -0.31 | Ga-Sc_b | Hf1P1_110 | -1.03 | Hf_t | Hf2Rh1_100 | -0.92 | Hf-Rh_b |
| Ga2Sc1_101 | -0.58 | Ga_t | Hf1P1_111 | -0.84 | Hf_t | Hf2Rh1_110 | -2.25 | HfHfHf_h |
| Ga2Sc1_110 | -0.19 | Ga-Ga_b | Hf1Ru1_001 | -1.09 | Hf-Ru_b | Ir1Ta1_001 | -3.40 | Ta_t |
| Ga2Sc1_111 | -0.81 | Sc_t | Hf1Ru1_100 | -1.09 | Hf-Hf_b | Ir1Ta1_101 | -1.32 | Ta_t |
| Ga2Th1_001 | -0.43 | Ga_t | Hf1Ru1_101 | -1.02 | Hf-hf_b | Ir1Ta1_111 | -0.55 | Ta-Ta_b |
| Ga2Th1_100 | -1.13 | GaThTh_h | Hf1Ru1_110 | -1.02 | Ru_t | Ir1V1_111 | -0.82 | Ir_t |
| Ge2Pt1_001 | -0.41 | Ge-Pt_b | Hf1Ru1_111 | -0.59 | Ru_t | Mg2Pt1_001 | -0.48 | Pt_t |
| Ge2Pt1_100 | -0.10 | Ge-Pt_b | Hf2Hg1_001 | -1.15 | Hf-Hf_b | Mg2Pt1_100 | -0.75 | Mg_t |
| Ge2Pt1_101 | -3.88 | Pt_t | Hf2Hg1_100 | -0.84 | Hg_t | Mg2Pt1_101 | -0.34 | MgMgMg_h |
| Ge2Pt1_110 | -0.32 | Pt_top | Hf2Hg1_101 | -1.18 | HfHgHg_h | Mg2Pt1_110 | -0.67 | Mg_t |
| Ge2Ti1_101 | -0.57 | GeGeTi_h | Hf2Hg1_110 | -1.03 | Hf-Hf-Hf_h | Mn1P1_001 | -0.38 | Mn-Mn_b |
| Ge2Ti1_110 | -1.61 | Ti_t | Hf2Hg1_111 | -1.27 | Hg_t | Mn1P1_100 | -0.85 | P_t |
| Hf1P1_001 | -0.33 | Hf_t | Hf2Ni1_101 | -1.18 | Hf-Hf-Hf_h | Mn1P1_101 | -0.43 | MnP_b |
| Hf1P1_100 | -1.47 | HfP_b | Hf2Ni1_110 | -0.98 | Hf-Hf-Hf_h | Mn1P1_110 | 0.75 | MnPP_h |
| Hf1P1_101 | -0.83 | Hf_t | Hf2Ni1_111 | -1.18 | Hf-Hf-Hf_h | Mn1P1_111 | -0.33 | P_t |
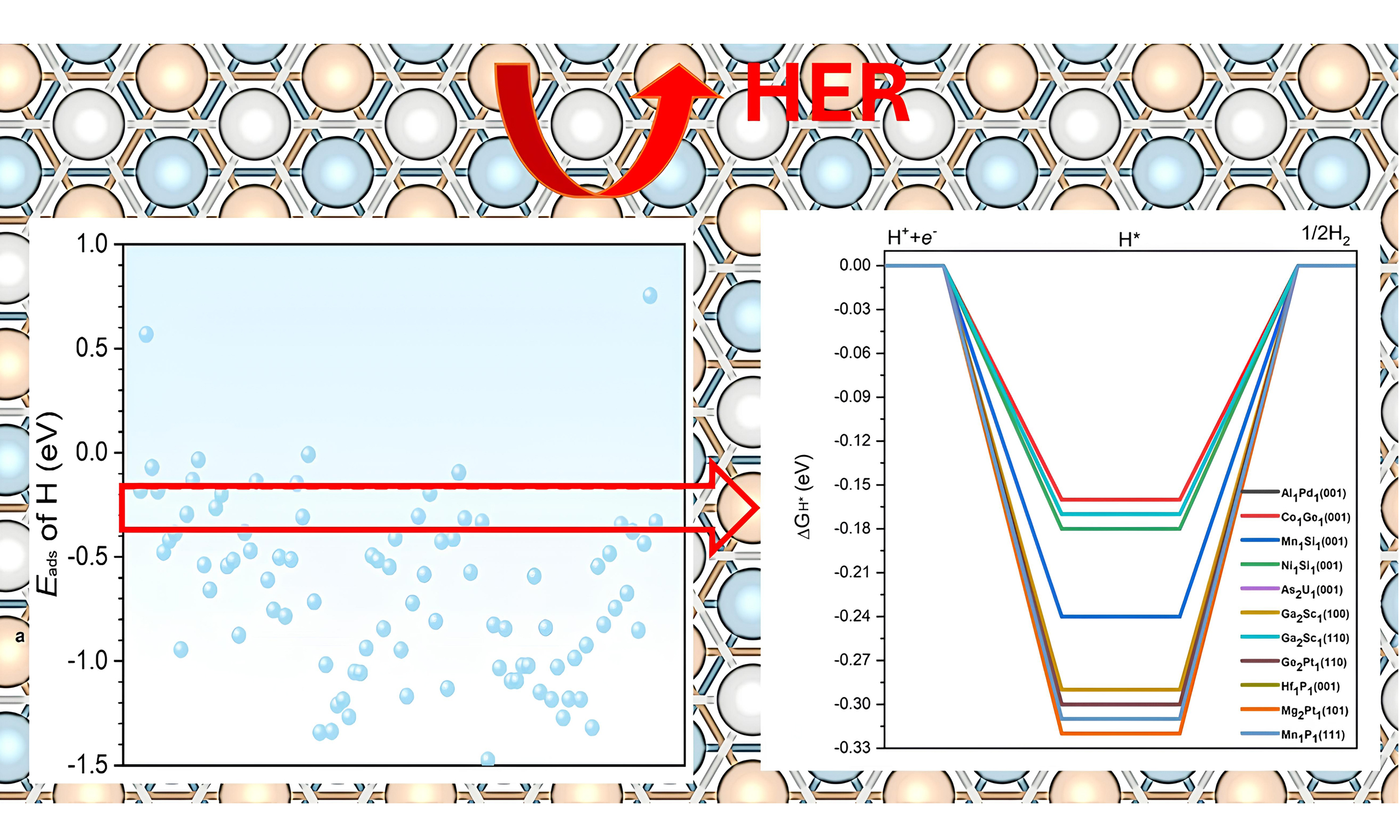
Statistical analysis shows that hydrogen adsorption energies on the surfaces of topological materials predominantly fall within -1.5 eV to 0 eV. As a result, we selected candidates whose Eads values satisfy these criteria. This focus was also motivated by the observation that the systems with Eads outside this range tend to bind hydrogen either too weakly or too strongly, making them unsuitable for further consideration. As summarized in Figure 2, the topological materials exhibit a well-distributed range of Eads values, highlighting their potential to provide abundant active sites. In addition, most of the selected topological materials possess a moderate adsorption capacity for hydrogen atoms, making them promising candidates for efficient HER.
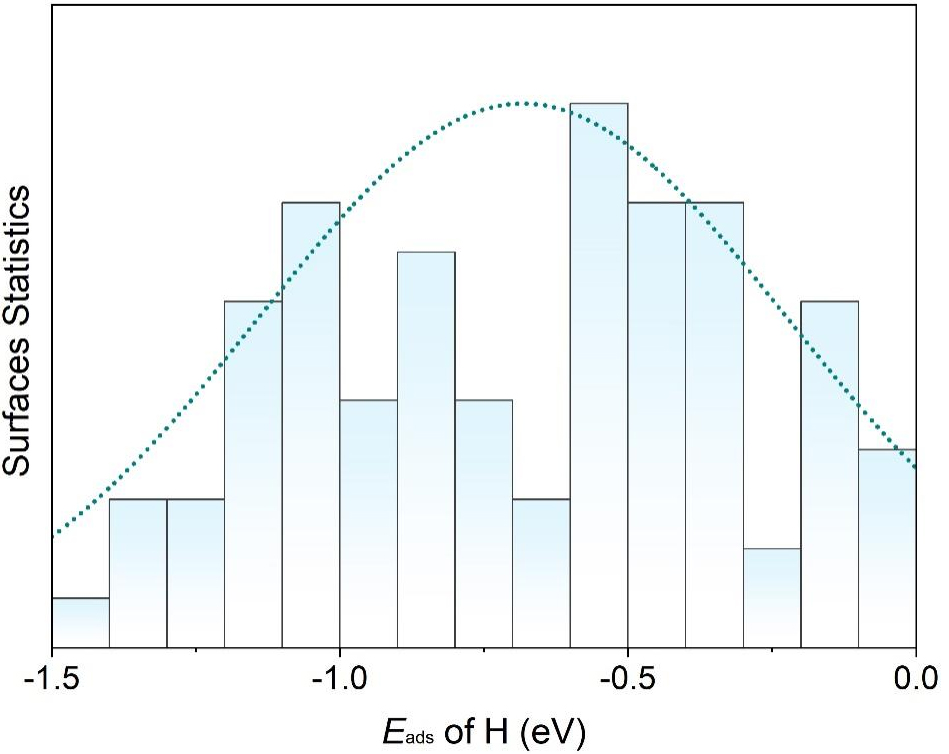
Figure 2. Normalized distribution of Eads (as in Table 1) for hydrogen on various topological materials, showing a well-distributed range and predominantly moderate hydrogen adsorption.
Preferred adsorption sites of the selected topological materials
It was reported that optimal activity and selectivity can be achieved when Eads of hydrogen is -0.27 eV[32,34]. This criterion is based on previous studies showing that adsorption energies close to -0.27 eV typically balance the hydrogen binding strength required for optimal HER activity. Grimme et al. demonstrated that adsorption energy near -0.27 eV is often ideal for achieving high HER activity across a variety of catalysts, providing a balance between adsorption and desorption processes[31]. Similarly, studies by Greeley et al. have reinforced that an optimal adsorption range enhances catalytic efficiency and stability[33]. With this criterion, we have pinpointed 11 distinct surfaces, with each displaying Eads of hydrogen within a 0.1 eV range of the optimal benchmark of -0.27 eV, as shown in Figure 3.
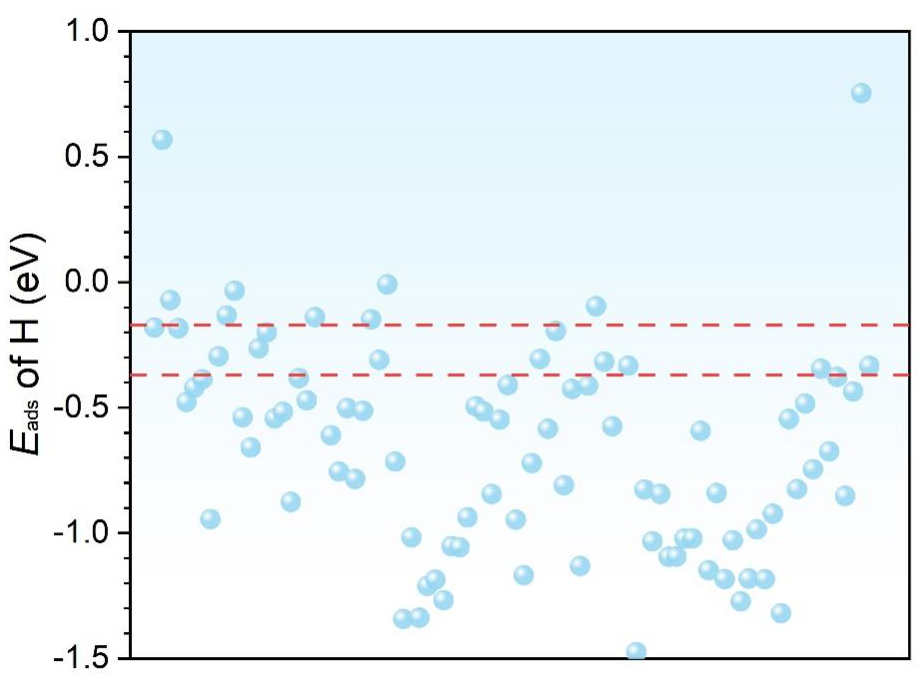
Figure 3. Adsorption energies of the most stable adsorption configurations of hydrogen atoms on different topological materials. The points in between the red dotted lines show those with Eads of hydrogen within the -0.27 ± 0.1 eV. As reported in the literature, the adsorption energies close to -0.27 eV typically balance the hydrogen binding strength required for optimal HER activity[31]. HER: Hydrogen evolution reaction.
After identifying the promising topological materials for HER, we systematically summarized the detailed atomic structures and adsorption behaviors of their surfaces, as illustrated in Figure 4. Interestingly, the favorable adsorption sites for hydrogen vary depending on both the material and the specific surface facet and can be the top, bridge, and hollow sites. This variation reflects the influence of local atomic arrangements, coordination numbers, and electronic environments on hydrogen binding. For example, hydrogen prefers Al-Pd bridge sites on Al1Pd1 (001) surface, where it can form optimal interactions with both neighboring atoms simultaneously, with the Eads calculated to be -0.18 eV. In contrast, hydrogen prefers Co-top sites with a slight tilt toward the neighboring Ge atom on the Co1Ge1(001) surface, with the Eadscalculated to be -0.18 eV. In the case of Ga2Sc1, both (100) and (110) serve as favorable hydrogen adsorption sites, but the preferred adsorption configurations differ between the facets. On the (100) surface, hydrogen binds most stably at the Ga-Sc bridge site, whereas on the (110) surface, the Ga-Ga bridge site is favored. The hydrogen adsorption energies on these two sites are calculated to be -0.31 eV and -0.19 eV, respectively. For the case of Ge2Pt1(110), hydrogen prefers Pt top sites with an Eads of -0.32 eV. These differences of the preferred adsorption sites highlight how subtle variations in surface atomic arrangement and termination can lead to distinct adsorption behaviors. The details of other geometries can be found in Figure 4. Overall, these observations highlight the critical importance of thoroughly examining multiple surfaces and adsorption sites for each material. Meanwhile, as expected, these topological materials offer robust surfaces and adsorption sites, which can accommodate hydrogen with optimal adsorption strength.
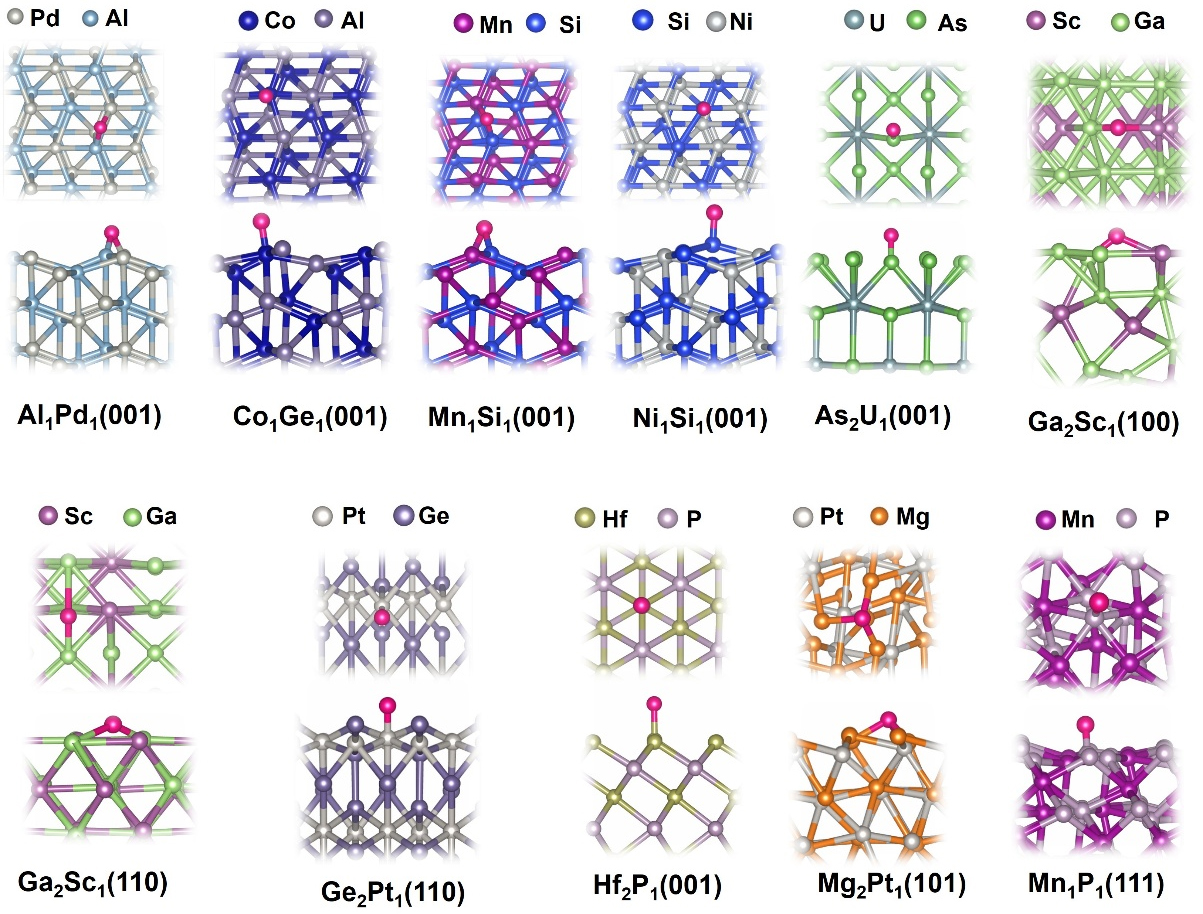
Figure 4. Top views and side views of the most stable hydrogen adsorption sites on 11 promising topological materials. The pink spheres represent hydrogen atoms, and the color schemes for other atoms are also provided for each topological material.
Gibbs free energy calculations
The ΔGH* is a well-accepted descriptor that has been successfully applied to predict HER activities[35]. Previous studies have established that an ideal ΔGH* close to zero is a strong indicator of effective HER activity, as it suggests a balanced adsorption and desorption process, which is critical for optimal hydrogen evolution[31,33]. Focusing on these 11 topological materials, we further calculated the ΔGH* and the results are summarized in Figure 5. It is seen that the ΔGH* values range from -0.16 eV to -0.32 eV, suggesting that these surfaces have the potential to function as high-performance catalysts for HER. Among these 11 topological materials,
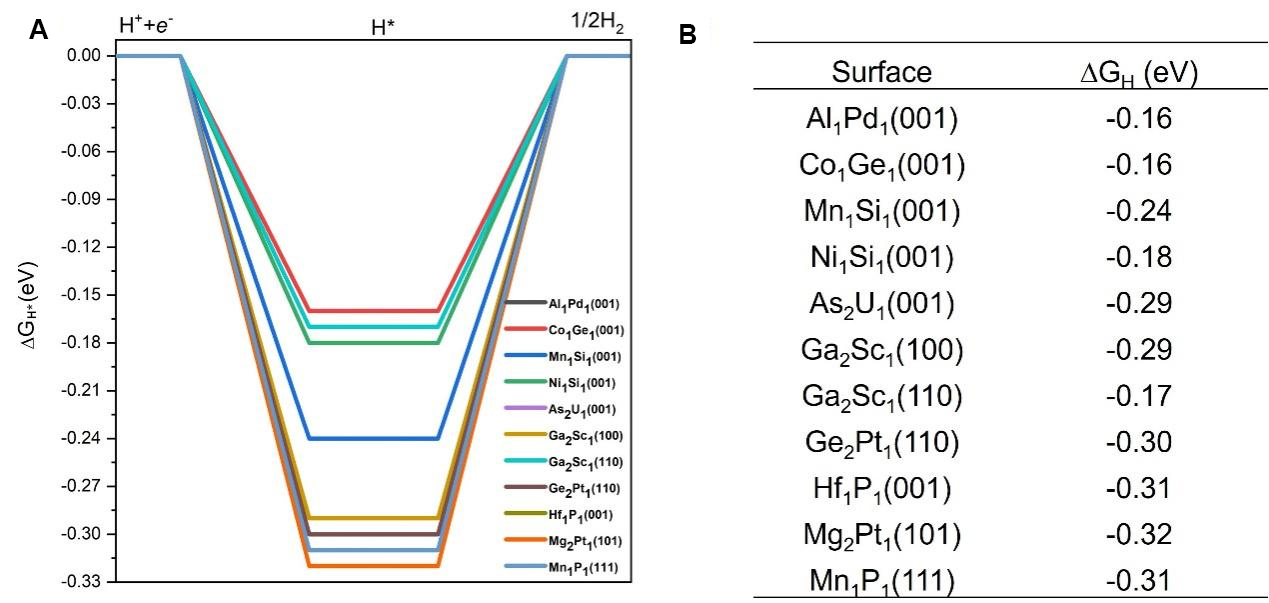
Figure 5. (A) DFT calculated free energy diagrams for HER on the 11 promising topological quantum materials with their corresponding surface termination. These eleven candidates are identified based on their optimal hydrogen adsorption energies, as discussed in Section 3.3. Some curves overlap due to similar ΔGH* values among different materials and surface terminations; (B) Summary table listing the corresponding adsorption surfaces and their calculated ΔGH* values. A ΔGH* value close to 0 eV is regarded as optimal for effective HER catalytic activity. HER: Hydrogen evolution reaction; DFT: density functional theory; ΔGH*: Gibbs free energy of hydrogen adsorption.
Discussion
Experimental studies on topological materials have demonstrated favorable catalytic properties that align well with our predictions, particularly regarding adsorption energies and ΔGH* values. For example, Qu et al. reported that Bi2Te3, a topological insulator, exhibited significant HER activity, achieving a current density of 1.74 μA cm-2, with partially oxidized surfaces contributing to enhanced catalysis[22]. Boukhvalov et al. demonstrated the HER activity of the topological nodal-line semimetal AuSn4, attributing its performance to unique surface states that facilitated electron transfer[24]. These findings highlight that topological materials can possess intrinsically advantageous properties for HER, supporting our predictions regarding adsorption sites and ΔGH* in the materials we studied.
In the present study, we selected an adsorption energy range of -0.27 ± 0.1 eV to evaluate HER performance. This criterion is based on previous studies that adsorption energies close to -0.27 eV typically balance the hydrogen binding strength required for optimal HER activity[31,33]. By adopting this criterion, our study provides a robust approach that can be replicated and built upon in future HER research involving topological materials.
Our study relies on the ΔGH* as a primary descriptor for assessing HER activity. While ΔGH* has been widely used as an adequate descriptor for evaluating HER performance, it may not fully capture the complex mechanisms of catalytic performance, especially in topological materials, where unique surface states and electronic properties are integral to their functionality[36]. In topological materials, the presence of surface states and high electron mobility could affect HER performance by facilitating charge transfer, enhancing catalytic activity even if ΔGH* values are suboptimal. Furthermore, reaction kinetics, including hydrogen diffusion rates on the catalyst surface, are essential for understanding HER mechanisms and can vary significantly in materials with complex electronic structures. In the future, electronic structure calculations, such as projected density of states (DOS) and Bader charge analysis, may be incorporated to further elucidate the charge transfer characteristics and reaction kinetics of the selected topological materials.
The DOS of 11 representative high-performance HER catalysts were calculated, and the corresponding d-band and p-band centers were extracted [Supplementary Figure 1]. Unlike conventional transition-metal catalysts, these topological materials do not exhibit a clear correlation between traditional electronic descriptors and HER performance. This deviation is likely attributed to the unique electronic characteristics of topological materials, such as nontrivial band topology, gapless surface states and surface Fermi arc states[36,37]. Clearly, a more in-depth exploration of unconventional electronic descriptors and their quantitative correlations with catalytic activity in these systems represents a fascinating direction for future research.
CONCLUSIONS
We conducted a comprehensive computational investigation of nearly 100 surfaces of topological materials to identify high-performance topological catalysts for efficient HER. We examined over 1,000 adsorption sites, including top, bridge, and various multi-fold sites. Our findings reveal that these topological materials exhibit a broad range of hydrogen adsorption energies, primarily ranging from -1.5 eV to 0 eV. Since previous studies suggested an optimal adsorption energy of -0.27 eV for ideal HER performance, we focused on materials with adsorption energies within -0.27 ± 0.1 eV. The computed Gibbs free energy ΔGH* values for these materials in the HER range from -0.31 eV to -0.16 eV, and detailed configurations for hydrogen adsorptions are provided. Ultimately, we identified 11 topological materials as promising catalysts for superior HER catalytic activity. Among these 11 topological materials, four candidates have ΔGH* very close to zero [Al1Pd1(001), Co1Ge1(001), Ni1Si1(001) and Ga2Sc1(110) with ΔGH* of -0.16, -0.16, -0.18 and -0.17 eV, respectively], indicating that they are likely to achieve the highest HER performance. These findings establish that topological materials possess abundant and diverse active sites, resulting in a wide range of hydrogen adsorption energies, thus enabling promising HER performance. These results can serve as a reliable dataset for further machine-learning studies and also offer valuable insights and guidelines for further theoretical and experimental exploration of topological materials as high-performance HER catalysts.
DECLARATIONS
Acknowledgments
The authors gratefully acknowledge the use of computing resources at the Agency for Science, Technology and Research (A*STAR) Computational Centre and the National Supercomputer Centre, Singapore.
Authors’ contributions
DFT calculations, formal analysis, writing, and review: Yang, J.
Formal analysis, review, and editing: Wu, Y.; Yu, Z.
Discussion and review: Politano, A.; Bukhvalov, D.; Cupolillo, A.
DFT calculations, formal analysis, and supervision: Feng, H.
Conceptualization and supervision: Zhang, X.; Zhang, Y. W.
Availability of data and materials
The raw data supporting the conclusions of this article are available from the authors upon reasonable request.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by the Italy-Singapore Science and Technology Cooperation (Grant No. R23101R040), the Singapore A*STAR SERC CRF Award, and the National Natural Science Foundation of China (22473007). Work at the University of L’Aquila and Calabria was funded by the European Commission - Next Generation EU, Mission 4 Component C2, Investment 1.1, under the Ministry of University and Research (MUR) of Italy PRIN 2022 (CUP: E53D23001750006, Grant No. 2022LFWJBR, acronym PLANET) and PRIN PNRR (CUP: E53D23018280001, Grant No. P20223LXTA, acronym ENTANGLE) projects. Politano, A.; Cupolillo, A.; Zhang, Y. W. acknowledge the EURIPIDES project, funded within the framework of the Italy-Singapore Science and Technology Cooperation 2023-2025 (Grant No. R23101R040 for A*STAR in Singapore and SG23GR07 for the Italian MAECI, Ministry of Foreign Affairs and International Cooperation).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Beasy, K.; Ajulo, O.; Emery, S.; Lodewyckx, S.; Lloyd, C.; Islam, A. Advancing a hydrogen economy in Australia: public perceptions and aspirations. Int. J. Hydrogen. Energy. 2024, 55, 199-207.
2. Koneczna, R.; Cader, J. Towards effective monitoring of hydrogen economy development: a European perspective. Int. J. Hydrogen. Energy. 2024, 59, 430-46.
3. Hassan, A.; Ilyas, S. Z.; Jalil, A.; Ullah, Z. Monetization of the environmental damage caused by fossil fuels. Environ. Sci. Pollut. Res. Int. 2021, 28, 21204-11.
4. Tiedje, J. M.; Bruns, M. A.; Casadevall, A.; et al. Microbes and climate change: a research prospectus for the future. mBio 2022, 13, e0080022.
5. San-Akca, B.; Sever, S. D.; Yilmaz, S. Does natural gas fuel civil war? Rethinking energy security, international relations, and fossil-fuel conflict. Energy. Res. Soc. Sci. 2020, 70, 101690.
6. Züttel, A.; Remhof, A.; Borgschulte, A.; Friedrichs, O. Hydrogen: the future energy carrier. Philos. Transact. A. Math. Phys. Eng. Sci. 2010, 368, 3329-42.
7. Kazemi, A.; Manteghi, F.; Tehrani, Z. Metal Electrocatalysts for hydrogen production in water splitting. ACS. Omega. 2024, 9, 7310-35.
8. Alasali, F.; Abuashour, M. I.; Hammad, W.; Almomani, D.; Obeidat, A. M.; Holderbaum, W. A review of hydrogen production and storage materials for efficient integrated hydrogen energy systems. Energy. Sci. Eng. 2024, 12, 1934-68.
9. Mei, L.; Zhang, Y.; Ying, T.; et al. Photochemical reduction of ultrasmall Pt nanoparticles on single-layer transition-metal dichalcogenides for hydrogen evolution reactions. Mater. Today. Energy. 2024, 42, 101487.
10. Chen, J.; Aliasgar, M.; Zamudio, F. B.; et al. Diversity of platinum-sites at platinum/fullerene interface accelerates alkaline hydrogen evolution. Nat. Commun. 2023, 14, 1711.
11. Smiljanić, M.; Panić, S.; Bele, M.; et al. Improving the HER Activity and stability of Pt nanoparticles by titanium oxynitride support. ACS. Catal. 2022, 12, 13021-33.
12. Peng, X.; Jin, X.; Gao, B.; Liu, Z.; Chu, P. K. Strategies to improve cobalt-based electrocatalysts for electrochemical water splitting. J. Catal. 2021, 398, 54-66.
13. Fang, W.; Wu, Y.; Xin, S.; et al. Fe and Mo dual-site single-atom catalysts for high-efficiency wide-pH hydrogen evolution and alkaline overall water splitting. Chem. Eng. J. 2023, 468, 143605.
14. Huo, L.; Jin, C.; Jiang, K.; Bao, Q.; Hu, Z.; Chu, J. Applications of nickel‐based electrocatalysts for hydrogen evolution reaction. Adv. Energy. Sustain. Res. 2022, 3, 2100189.
15. Shao, W.; Xing, Z.; Xu, X.; et al. Bioinspired proton pump on ferroelectric HfO2-coupled Ir catalysts with bidirectional hydrogen spillover for pH-universal and superior hydrogen production. J. Am. Chem. Soc. 2024, 146, 27486-98.
16. Yang, J.; Yu, Z. G.; Zhang, Y. W. Synergizing Cu dimers and N atoms in graphene towards an active catalyst for hydrogen evolution reaction. Nanoscale. Adv. 2021, 3, 5332-8.
17. Zheng, Y.; Xiao, S.; Xing, Z.; et al. Oxophilic vanadium and deprotonated ruthenium atoms on tungsten carbide with accelerated intermediate migration for high-performance seawater hydrogen evolution. Nano. Energy. 2024, 127, 109769.
18. Zheng, Y.; Geng, W.; Xiao, S.; et al. Interfacial Ir-V direct metal bonding enhanced hydrogen evolution activity in vanadium oxides supported catalysts. Angew. Chem. Int. Ed. 2024, 63, e202406427.
20. Kumar, N.; Guin, S. N.; Manna, K.; Shekhar, C.; Felser, C. Topological quantum materials from the viewpoint of chemistry. Chem. Rev. 2021, 121, 2780-815.
21. Luo, H.; Yu, P.; Li, G.; Yan, K. Topological quantum materials for energy conversion and storage. Nat. Rev. Phys. 2022, 4, 611-24.
22. Qu, Q.; Liu, B.; Liang, J.; et al. Expediting hydrogen evolution through topological surface states on Bi2Te3. ACS. Catal. 2020, 10, 2656-66.
23. Li, J.; Ma, H.; Xie, Q.; et al. Topological quantum catalyst: dirac nodal line states and a potential electrocatalyst of hydrogen evolution in the TiSi family. Sci. China. Mater. 2017, 61, 23-9.
24. Boukhvalov, D. W.; D'Olimpio, G.; Mazzola, F.; et al. Unveiling the catalytic potential of topological nodal-line semimetal AuSn4 for hydrogen evolution and CO2 reduction. J. Phys. Chem. Lett. 2023, 14, 3069-76.
25. Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 1996, 54, 11169.
26. Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B. 1994, 49, 14251.
27. Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865-8.
29. Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B. 1999, 59, 1758-75.
30. Petrilli, H. M.; Blöchl, P. E.; Blaha, P.; Schwarz, K. Electric-field-gradient calculations using the projector augmented wave method. Phys. Rev. B. 1998, 57, 14690-7.
31. Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104.
32. Nørskov, J. K.; Bligaard, T.; Logadottir, A.; et al. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, J23.
34. Greeley, J.; Jaramillo, T. F.; Bonde, J.; Chorkendorff, I. B.; Nørskov, J. K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909-13.
35. Liu, X.; Xiao, J.; Peng, H.; Hong, X.; Chan, K.; Nørskov, J. K. Understanding trends in electrochemical carbon dioxide reduction rates. Nat. Commun. 2017, 8, 15438.
36. Meng, W.; Zhang, X.; Liu, Y.; et al. Multifold fermions and fermi arcs boosted catalysis in nanoporous electride 12CaO·7Al2O3. Adv. Sci. 2023, 10, e2205940.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.