

Multiscale insights into the interaction and reaction of water at solid surfaces from theoretical simulations
 0
0 Abstract
Solid-water interfaces play critical roles in environmental, energy, and catalytic processes, yet their complex multiscale behaviors remain challenging to fully understand. This review systematically examines the interaction and reaction mechanisms of water at solid surfaces through a hierarchical theoretical simulation framework - spanning atomic, mesoscopic, and macroscopic scales. At the atomic level, we discuss defect engineering and electronic reconstruction strategies that govern water adsorption and activation. At the mesoscale, we explore collective dynamics, including hydrogen-bond networks and nanoconfinement effects, which influence proton transport and solvation structures. At the macroscopic level, we analyze how external fields (electric, light, chemical) modulate interfacial processes and reaction pathways. By integrating multiscale simulations from density functional theory to molecular dynamics simulation, this review bridges the gap between molecular insights and system-level performance, offering a predictive foundation for the rational design of advanced interfacial materials in energy conversion, environmental remediation, and ionic devices.
Keywords
INTRODUCTION
Chemical reactions at the solid-water interface lie at the heart of regulating biogeochemical cycles and environmental processes, a concept deeply rooted in the foundational theories developed within interface science[1]. As early as the twentieth century, Irving Langmuir pointed out that solid-phase chemical reactions are inherently surface phenomena, with surface structure playing a decisive role[2]. In the 1980s, a breakthrough in experimental methodology - synchrotron-based X-ray absorption spectroscopy - enabled, for the first time, in situ observation of the molecular structure of inner-sphere (IS) complexes formed by anions in real aqueous environments[3]. Subsequent systematic studies have elucidated the complex interplay among metal oxides, aqueous solutions, and microorganisms, while also highlighting the challenge of the “pressure gap” between model systems under ultra-high vacuum and real aqueous environments[4].
On the theoretical front, the development of the charge distribution multisite complexation model directly correlates surface protonation and ion adsorption behavior with crystal structure, thereby establishing a quantitative thermodynamic framework for predicting interfacial reactions[5]. Meanwhile, models advanced through crystal chemistry and solvation theory have bridged the intrinsic properties of minerals with their macroscopic interfacial electrical characteristics[6]. Lee et al. employed X-ray reflectivity to capture the exchange dynamics of monovalent cations at the mica-water interface[7]. Their work revealed a non-classical competitive adsorption behavior that cannot be accounted for by the classical electric double-layer theory[7]. Recently, the properties of nanoconfined interfacial water have also bridged a vital connection between classical interface science and emerging disciplines. For instance, Fumagalli et al. discovered that the dielectric constant of water plummets within sub-nanometer confined spaces[8], reshaping the understanding of ion transport through nanochannels. In solid-water interfacial chemistry, water serves multiple critical functions: it acts not only as a solvent medium, but also undergoes dissociative adsorption to form reactive surface sites[9], thereby regulating interfacial acidity and reactivity[10]. Through Grotthuss-type proton-transfer networks, it enables rapid and dynamic equilibration of surface charge[11,12]. Under nanoconfinement, its significantly altered structure and dynamics[13] can enhance ion adsorption and shift reaction pathways[14]. Simultaneously, as the primary component of ionic hydration shells, the orientation and hydrogen-bonding network of water molecules govern the interfacial distribution, adsorption free energy, and transport behavior of ions[15]. These roles are tightly coupled, collectively dictating the complex behavior of ions at the interface.
Solid-water interfaces are of significant applied value in critical fields such as chemistry, energy, environmental science, and ionic devices. This importance stems from their unique and tunable properties, including surface charge characteristics, complexation reaction mechanisms, and structural dynamics [Figure 1]. In the field of environmental science, for example, researchers have constructed MnO2-based catalysts featuring oxygen vacancies with distinct symmetries[16]. Additionally, a recent study demonstrates that photoexcited holes can drive the formation of surface high-valent cobalt-oxo species on a Co3O4/BiVO4 catalyst, utilizing water as the oxygen atom source for efficient pollutant degradation, further highlighting the critical role of solid-water interfaces in environmental remediation[17]. In the field of energy, the construction of a Zr-O-W interface on a Pt single-atom-modified ZrO2-WO3 heterojunction has been employed to address the challenges of slow proton supply kinetics and the high energy barrier for C-C coupling in light-driven CO2 reduction[18]. Alternatively, an opposite strategy for aqueous energy storage involves suppressing parasitic water decomposition at the electrode interface, where the use of glycine as an electrolyte additive achieves a record discharge voltage of 1.83 V in magnesium-air batteries[19]. In the field of ionic devices, high-performance bioinspired systems can be engineered by precisely tuning the geometric structure, surface charge, and chemical properties of nanochannels[20-21]. In summary, targeted regulation of solid-water interfaces serves as a cornerstone for addressing complex environmental challenges, advancing sustainable energy systems, and overcoming performance limitations in ionic devices.
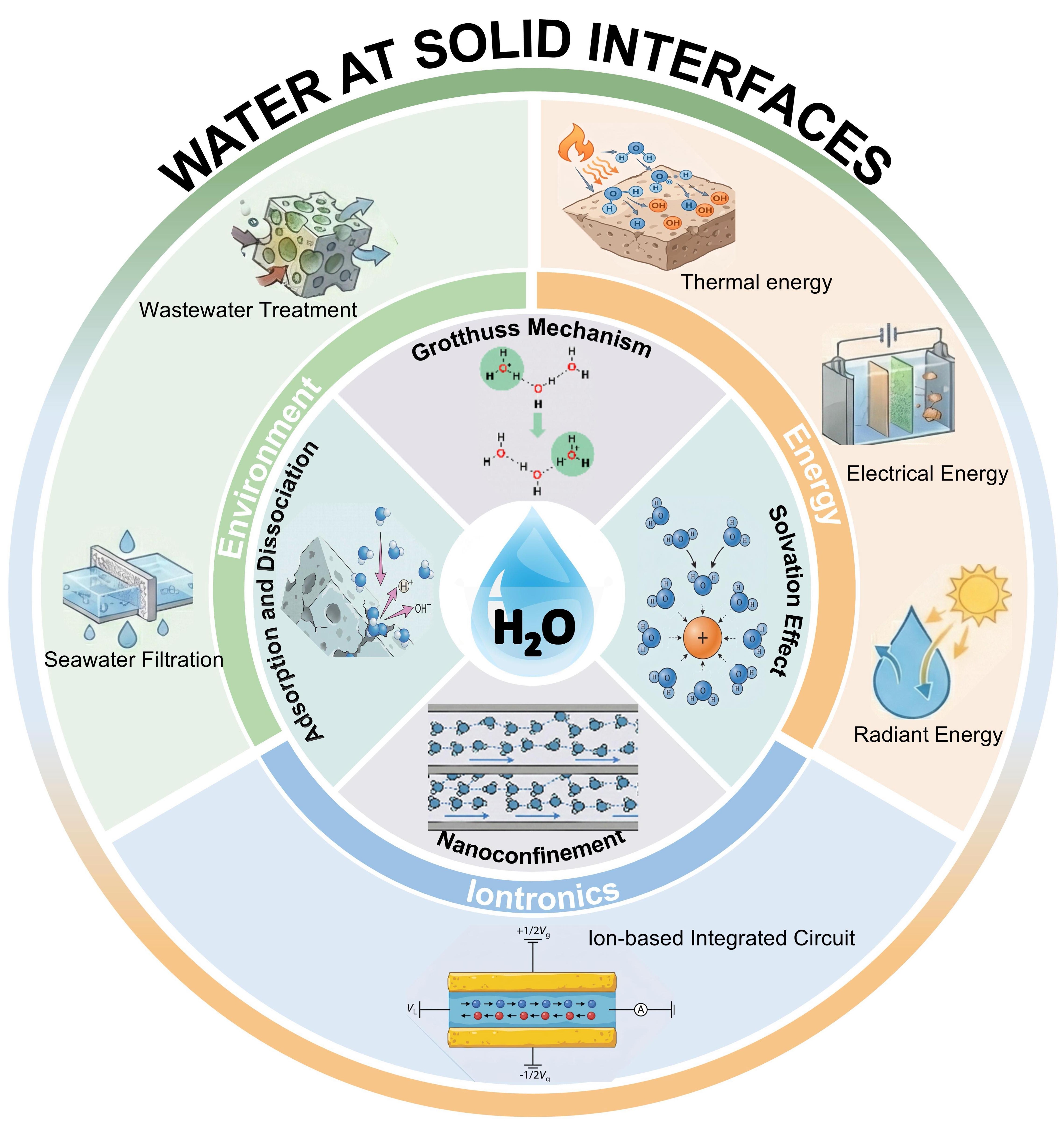
Figure 1. Solid-water interfacial systems and their typical applications.
As the critical microenvironment for ion adsorption, transport, and reaction, dynamic processes at the solid-water interface span timescales from femtoseconds to seconds. Traditional experimental techniques encounter significant limitations in capturing transient ion configurations, rapid proton transfer, and short-lived intermediates. For instance, while surface X-ray scattering techniques such as crystal truncation rod (CTR) and resonant anomalous X-ray reflectivity (RAXR) are effective in resolving static adsorption structures[10], their temporal resolution is often insufficient for tracking dynamic ion-exchange processes[7]. Meanwhile, in situ spectroscopic methods such as attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy can be employed to monitor adsorption kinetics[22]. However, they still lack sufficient spatial resolution and real-time observational capability to probe interfacial behavior under conditions of low ion concentration or nanoconfinement[23,24]. These limitations often render the solvation structure, transient coordination states, and charge-transfer pathways of interfacial ions largely inaccessible to direct observation, creating a mechanistic “black box”. Advances in theoretical calculations and multiscale simulations provide a vital pathway to address these limitations. Density functional theory (DFT) calculations and ab initio molecular dynamics (AIMD) simulations can reveal, at the atomic scale, the electronic structure characteristics of ion adsorption and their dynamic solvation environments[25]. This enables the prediction of acidity constants for interfacial proton transfer[26] and clarifies anomalous diffusion behavior of water and ions within nanopores[27]. Furthermore, methods such as molecular dynamics (MD) simulations extend the accessible temporal and spatial scales of modeling, which allows for the quantitative prediction of complex processes such as charge transport[28] and ion exchange[29]. These computational tools not only bridge gaps in experimental observation but also provide a mechanistic foundation and guidance for designing novel interfacial materials and optimizing environmental remediation strategies.
In summary, significant progress has been made in recent years in understanding the mechanistic, structural, and functional complexity of solid-water interfacial systems. However, substantial challenges remain in comprehending their microscopic mechanisms and multiscale correlations. In light of this, this review focuses on theoretical simulation studies, constructing a cross-scale framework from the atomic to the macroscopic level [Figure 2]. The specific research scope is outlined as follows: First, at the atomic/molecular scale (Å-nm), we systematically elucidate the underlying mechanisms of static bonding characteristics, electronic structure properties, and dynamic solvation processes between ions and surface sites. At the mesoscale (nm-μm), we delve into the collective diffusion behavior, competitive adsorption mechanisms, and interfacial transport kinetics of ions within hydrogen-bond networks (HBNs) and nanoconfined environments. Ultimately, at the macroscopic scale (μm-m), we clarify how these microscopic processes govern colloidal stability, contaminant transport, reactive transport, and overall system performance, considering factors such as electric field modulation, photocatalytic effects, and molecular competition. These distinct scales are interconnected through the parameterization of mechanisms and the coupling of models. The core objective is to bridge the two major bottlenecks of “chemical complexity” and “scale disparity”, thereby providing a systematic theoretical blueprint and technical foundation for predicting and precisely manipulating interfacial ion behavior in real environmental and engineered systems.
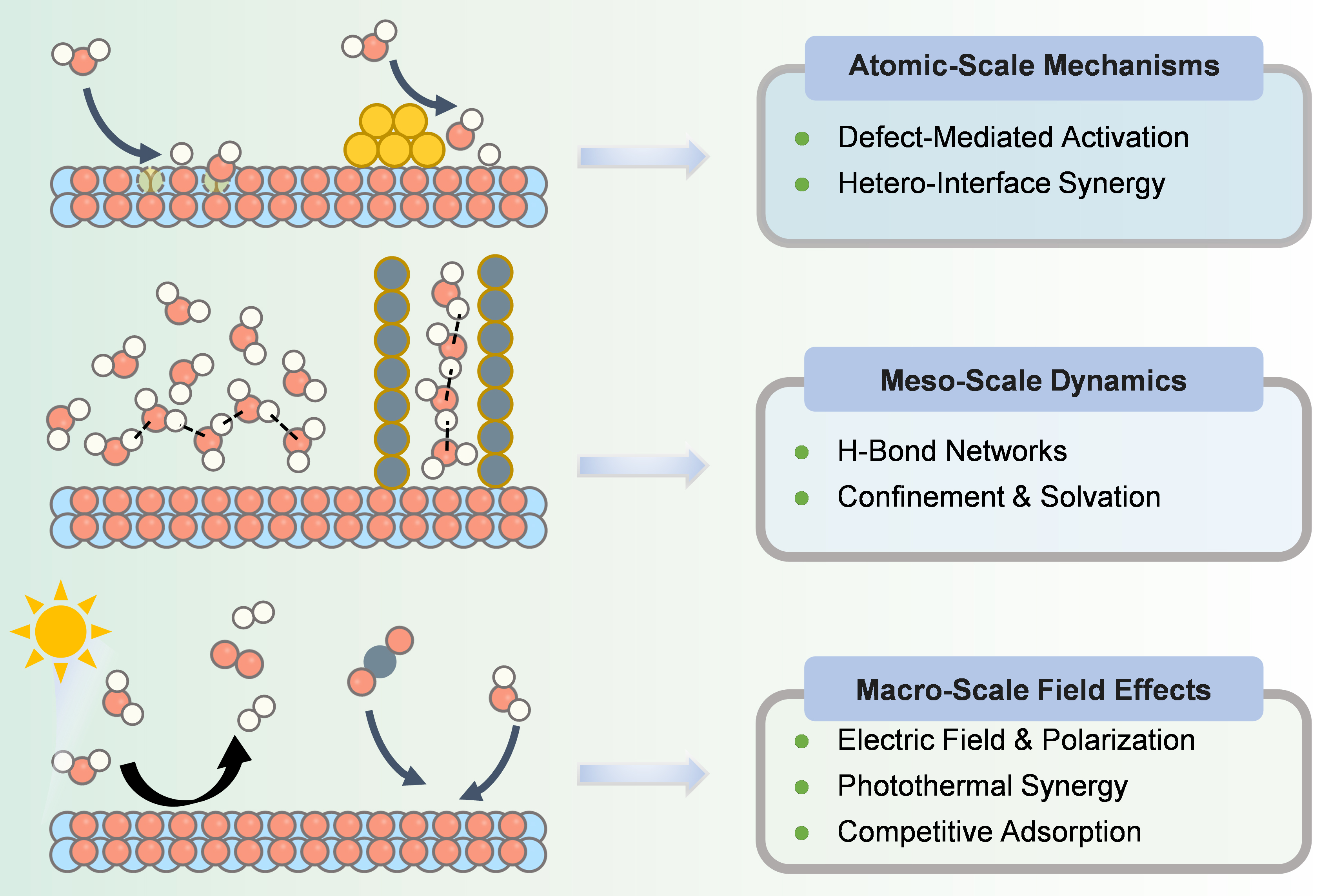
Figure 2. Multiscale insights into water-solid interfaces: from atomic/molecular through mesoscale to macroscopic levels.
ATOMIC-SCALE INTERACTION MECHANISMS BETWEEN WATER AND SOLID SURFACES
The water-solid interface is characterized by pronounced entropic complexity and intrinsic multiscale features. Although macroscopic fluid transport is governed by the coupling between cohesive forces and interfacial interactions, the physicochemical properties of such systems fundamentally originate from microscopic mechanisms: interatomic and intermolecular interactions at the atomic scale govern wetting behavior, while the local nanoscale environment directly determines interfacial binding strength and reactivity[30].
Despite the fact that real interfacial reactions involve complex solvation networks, the thermodynamic feasibility of water molecule adsorption and activation largely depends on the geometric configuration and electronic structure of local active sites[31]. At the atomic scale, ideal crystal lattices, owing to the translational symmetry of their electronic structures and their intrinsic chemical inertness, often struggle to overcome the high dissociation barrier of the O-H bond in water molecules[32]. Therefore, the construction of efficient water-solid interfaces critically relies on overcoming this intrinsic reaction inertness. This section focuses on two primary atomic-scale regulation strategies: (i) intrinsic defect engineering, which elucidates how oxygen vacancies reduce O-H bond dissociation barriers and alter water adsorption modes, as well as how surface hydroxyls evolve from reaction products into active catalytic species; and (ii) heterogeneous interface design, which highlights the synergistic activation of water molecules enabled by precise regulation of single-atom coordination environments and the “perimeter effect” at metal-oxide interfaces.
Capture and activation at defect sites
Idealized stoichiometric crystal surfaces, owing to their highly saturated electronic structures, typically exhibit a limited capability for water molecule activation. In contrast, real material surfaces are generally rich in lattice defects. By breaking the translational symmetry of the crystal lattice, these defects introduce coordinatively unsaturated sites, giving rise to localized electronic states and pronounced modulation of the local density of states (LDOS)[33]. Such defect sites not only enhance the Lewis acidity of surface metal centers but also induce local charge redistribution and the formation of microscopic electric fields. These effects exert strong polarization on adsorbed water molecules, substantially weakening the O-H bond strength and lowering the corresponding dissociation barrier[34]. Consequently, the reactivity of the water-solid interface is fundamentally governed by defect-dominated reconstruction of the potential energy surface. This section focuses on two key types of defects - oxygen vacancies acting as electron traps and surface hydroxyls functioning as proton-transfer nodes - to elucidate their decisive roles in water capture and activation.
The critical role of oxygen vacancies
As one of the most prevalent anionic defects on solid surfaces, the essential role of oxygen vacancies originates not merely from local geometric incompleteness, but from their profound reshaping of surface electronic-state distributions. Extensive theoretical and experimental studies have demonstrated that oxygen vacancies often serve as electron-rich active centers capable of markedly altering water adsorption configurations, enhancing interfacial electron-transfer capability, and thereby reshaping water activation pathways and subsequent reaction routes.
In typical transition-metal oxide systems, oxygen vacancies exhibit dual characteristics as both “electron traps” and “reactive sites.” Wang et al. employed hydrogen reduction of fluorine-doped TiO2 nanosheets (F-TiO2-x) to induce and stabilize surface oxygen vacancies, thereby significantly enhancing the photocatalytic degradation efficiency of indoor volatile organic compounds (VOCs)[35]. DFT calculations elucidated that the introduction of oxygen vacancies increased the adsorption energy of water molecules from 0.727 eV to 1.257 eV and provided key active sites that facilitate electron transfer to adsorbed water molecules (Δq = -0.25 e) [Figure 3A]. This process substantially lowered the reaction barrier for water dissociation to generate ·OH radicals, thereby establishing a reaction pathway in which surface fluorine and oxygen vacancies cooperatively promote water activation. Similarly, Li et al. combined DFT calculations with X-ray absorption fine structure (XAFS) spectroscopy to demonstrate that oxygen vacancies on the BiOCl (010) facet induce dissociative water adsorption[39].
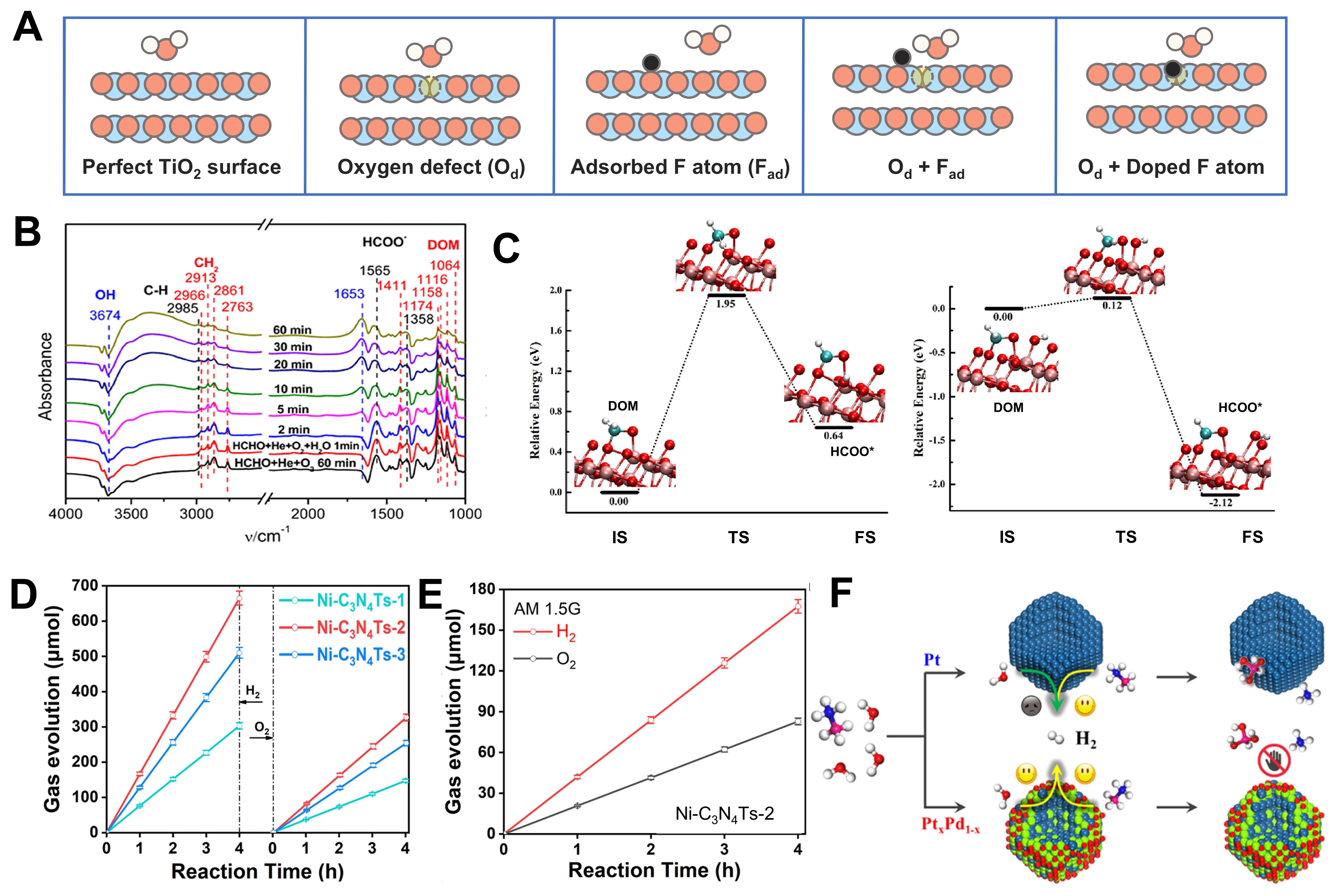
Figure 3. (A) Adsorption and activation of water molecules on surfaces; (B) In situ DRIFTS showing the dynamic evolution of TiO2 samples under a formaldehyde flow for 60 min; (C) Distinct reaction pathways for DOM conversion to formate: H transfer from DOM to a surface lattice oxygen species (left) versus H transfer to a surface terminal hydroxyl group (right). (B and C) Ref.[36] Copyright © 2020 American Chemical Society; (D) Overall water-splitting activity of Ni-C3N4Ts under visible-light irradiation; (E) Overall water-splitting activity of Ni-C3N4Ts-2 under a standard solar simulator. (D and E) Ref.[37] Copyright © 2022 American Chemical Society; (F) Schematic illustration of hydrogen production activity and durability based on monometallic Pt/CNT and bimetallic PtxPd1-x/CNT catalysts. Ref.[38] Copyright © 2020 American Chemical Society. DRIFTS: Diffuse reflectance infrared Fourier transform spectroscopy; DOM: dioxymethylene; CNT: carbon nanotube.
However, oxygen vacancies do not universally exhibit strong activation effects across all material systems. For example, on the WO3-x (001) surface, the introduction of oxygen vacancies triggers pronounced surface reconstruction, causing originally undercoordinated W sites to evolve toward geometric and electronic configurations similar to the W5c sites on stoichiometric surfaces. This reconstruction-induced “defect healing” effect thermodynamically stabilizes the surface structure but simultaneously weakens the defect’s ability to activate reactants[40]. Computational results show that the adsorption energy of water molecules on the defective surface (1.05 eV) differs only marginally from that on the pristine surface (0.95 eV), with water molecules favoring molecular adsorption rather than dissociation[40]. This indicates that defect-induced water activation can be significantly constrained by lattice reconstruction and stabilization mechanisms.
Taken together, these observations indicate that the essence of oxygen-vacancy engineering lies not in a simple increase in defect concentration, but rather in the precise regulation of defect-induced electronic states and local structural evolution. Rationally stabilized oxygen vacancies can effectively reduce the energy barriers associated with water activation and electron transfer, whereas excessive structural relaxation and coordination reconstruction may lead to defect passivation or even functional failure. Moreover, the role of oxygen vacancies extends beyond initial adsorption to include the dynamic modulation of proton supply during subsequent reaction steps.
The dual function of surface hydroxyls
Recent studies have reshaped the conventional understanding of surface hydroxyls. Rather than being merely passive products of water dissociation or inert adsorption sites, surface hydroxyls are now widely recognized as key active species governing the thermodynamics and kinetics of catalytic reactions at the water-solid interface. Extensive in situ spectroscopic investigations and theoretical calculations have demonstrated that surface hydroxyls can directly participate in proton-coupled electron transfer (PCET) processes and, through the construction of ordered interfacial HBNs, significantly lower the activation barrier associated with the rate-determining step[36,41].
Chen et al. combined in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) with DFT calculations to elucidate the promoting role of surface hydroxyls in the conversion of dioxymethylene (DOM) to formate[36]. Mechanistic calculations identified two distinct reaction pathways[36]: H transfer from DOM to a surface lattice oxygen species involves a high activation barrier of 1.95 eV, whereas H transfer to a surface terminal hydroxyl requires a barrier of only 0.12 eV and proceeds exothermically. Beyond direct participation in reaction pathways, surface hydroxyls and interfacial water structures can also indirectly regulate reaction kinetics through collective hydrogen-bonding effects. Liu et al. combined in situ Raman spectroscopy with AIMD simulations to show that the accumulation of high-density surface holes induces the rearrangement of interfacial water molecules, forming highly ordered HBNs[41]. In this system, adjacent Fev=O intermediates activate interfacial adsorbed water molecules - rather than hydroxide ions - via hydrogen bonding, enabling a multi-electron reaction pathway with an almost negligible activation barrier[41].
In summary, interfacial water molecules play a dual functional role: dissociated water generates surface hydroxyls that provide low-barrier proton-transfer channels, while intact water molecules assemble into ordered HBNs that cooperatively promote efficient substrate activation.
Electronic reconstruction at single-atom and bimetallic interfaces
The efficiency of charge transport at solid-liquid interfaces, particularly the orbital interactions between solid surfaces and solvent water molecules, often dictates interfacial reaction rates. Consequently, regulating the electronic structure of active sites to optimize water adsorption and activation has emerged as an effective strategy for enhancing catalytic performance. In single-atom catalysts (SACs) and bimetallic or interfacial systems, electronic reconstruction induced by strong metal-support interactions (SMSI) plays a decisive role.
On one hand, coordination between single metal atoms and substrates modifies local electronic states and improves orbital matching with the frontier molecular orbitals of water molecules[42]. On the other hand, the interfacial “perimeter effect” induces charge transfer due to differences in work function, establishing a local electric field[43]. These changes in the electronic microenvironment effectively reshape the interfacial configuration of water molecules and lower the dissociation barrier of the O-H bond in water molecules. This section aims to provide an in-depth analysis of the electronic reconstruction mechanisms in these two systems and their regulatory roles in solid-liquid interfacial reaction activity.
Coordination environment of single atomic sites
At the atomic scale, the electronic structure of metal active centers exhibits high tunability regarding the local coordination environment. Evolving from isolated single atoms to sub-nanometer metal clusters, parameters such as coordination number, ligand electronegativity, and bond length serve as important descriptors for regulating interfacial electronic reconstruction[44]. SACs are firmly anchored through the formation of strong covalent or ionic bonds with substrate heteroatoms (such as O, N, S). Due to the lack of homonuclear metal-metal bonds, they exhibit significant charge transfer and electron-deficient (cationic) characteristics[45]. The evolution of electronic structures under these different coordination configurations is crucial for revealing the flipping of water molecule adsorption configurations at the solid-liquid interface and the mechanism by which the d-band center regulates the dissociation barrier of the O-H bond.
Huang et al. developed an entropy-driven strategy for single-atom photocatalyst design by confining Ni single atoms within an interlayer structure via a hydrothermal method[37], which can induce and strengthen an internal macroscopic polarization electric field (PEF). Driven by the enhanced PEF, photogenerated charge carriers overcome Coulombic attraction and are efficiently separated and directionally transported to surface Ni-(s-triazine)4 active sites through channels formed by Ni-N bonds[37]. Su et al. employed an aqueous-phase reforming strategy to construct a hydrophilic hydroxylated Ru single-atom interface (HO-Ru/TiN) on a metallic TiN substrate[46]. Surface-bonded hydroxyls were shown to participate in the formation of unique HO-RuN5-Ti Lewis acid-base pairs, which not only improved interfacial wettability but also acted as active centers for efficient water capture and dissociation. Detection of OH and OOH intermediates by in situ spectroscopy confirmed that water molecules are oxidized by photogenerated holes at the interface to release O2 and supply the protons and electrons required for CO2 reduction, thereby establishing an interfacial hydroxyl-mediated water-solid synergistic catalytic mechanism.
Overall, regulation of the single-atom coordination environment provides an effective pathway for optimizing water-solid interfacial interactions. By constructing specific coordination configurations, such as confined architectures or surface hydroxylation, catalyst surface polarization and wettability can be enhanced, promoting water chemisorption and activation[46]. This strengthened interfacial coupling facilitates charge injection to the surface and effectively lowers the O-H bond dissociation barrier, thereby improving reaction kinetics.
The “perimeter effect” at metal-oxide interfaces
Isolated metal surfaces often exhibit limited capability for water activation due to weak metal-water interactions, which hinder the simultaneous stabilization of protonic and hydroxyl intermediates, resulting in kinetically constrained water dissociation[47]. In contrast, when metal nanoparticles are supported on oxide substrates, the interfacial regions formed between the two components, commonly referred to as perimeter sites, frequently display interfacial chemical properties distinct from those of either component alone[48]. These interfaces not only reconstruct local electronic structures and electric-field distributions but, more importantly, introduce a dual-site synergistic adsorption mode: oxide sites preferentially stabilize hydroxyl species (OH*), whereas metal sites favor the adsorption of protons or hydrogen atoms (H*). Such spatial and electronic cooperation lowers O-H bond cleavage barriers, rendering water dissociation both thermodynamically favorable and kinetically accessible[48].
Chen et al. engineered highly controllable Pt-PdO perimeter sites on carbon nanotube supports via atomic-scale interface design, achieving simultaneous enhancement of catalytic activity and stability[38]. MD simulations combined with DFT calculations revealed that Pd atoms preferentially segregate to nanoparticle surfaces and undergo partial oxidation under thermodynamic driving forces, ultimately forming a Pt-rich core-PdO-rich shell heterostructure. At the Pt-PdO interface, a pronounced bifunctional synergistic effect was observed [Figure 3F]: PdO sites substantially reduced the activation barrier for water dissociation from 0.83 eV to 0.11 eV, thereby accelerating the rate-determining step. In a classical model system, Fujitani et al. investigated Au/TiO2(110) and directly observed water dissociation at the junction between gold nanoparticles and the TiO2 support, yielding surface H and OH species[49]. This experimental evidence conclusively confirms that metal-oxide perimeter sites, rather than isolated metal or oxide surfaces, serve as the key active centers for water activation and subsequent surface reactions.
In other words, the perimeter effect introduced by metal-oxide interfaces fundamentally reshapes water adsorption and dissociation pathways on solid surfaces. By breaking the adsorption-dissociation equilibrium constraints inherent to single-component surfaces through interfacial synergy, these interfaces continuously generate highly reactive hydroxyl or hydrogen species.
MESOSCALE COLLECTIVE DYNAMICS OF WATER AT SOLID SURFACES
In practical systems, water reactions on solid surfaces often go beyond static descriptions of isolated active sites and instead involve the dynamic behavior of water molecular network at the mesoscale. At this scale, the connectivity of hydrogen-bond (HB) networks, orientational anisotropy, and the reconfigurability of solvation shells collectively determine proton and ion migration pathways and the overall thermodynamic driving forces[50]. This section systematically elucidates how surface HBNs and confined solvation structures transcend single-molecule models and thereby govern macroscopic catalytic and electrochemical performance.
Surface hydrogen-bond networks and proton transport
At the water-solid interface, water molecules are not randomly stacked. Instead, they are constrained by the local electric field of the solid surface and surface functional groups, forming an interfacial HB network with a certain degree of order through directional hydrogen bonding interactions[51]. This network typically exhibits a layered structure and restricted rearrangement dynamics. Its connectivity and stability directly determine the continuity and efficiency of proton migration along hydrogen-bonded chains via the Grotthuss mechanism[52]. Therefore, the structural characteristics and dynamic behavior of the interfacial water hydrogen bond network serve as a fundamental basis for understanding water-solid interfacial interactions.
Proton shuttling
Proton transport at the water-solid interface predominantly follows the Grotthuss mechanism, in which charge migration proceeds through the concerted breaking and formation of O-H bonds within a HBN, rather than via the physical diffusion of hydrated protons[53]. The efficiency of this mechanism under interfacial conditions critically depends on whether the HBN maintains long-range connectivity and orientational coherence[54]. At water-solid interfaces, however, the solid surface potential can disrupt the cooperative orientation of interfacial water molecules, leading to fragmentation or localization of HBNs and raising questions regarding the viability of efficient proton shuttling[55]. Recent in situ characterizations and AIMD simulations demonstrate that, as long as HB channels spanning multiple water molecules are preserved, the Grotthuss mechanism not only persists at interfaces but can even be significantly amplified under specific conditions[56].
Cai et al. computationally compared two proton-transfer pathways on the ZrO2(111) surface in aqueous environments[57]. They showed that direct proton hopping between surface oxygen sites (O2c to O3c) encounters an energy barrier as high as 128 kJ mol-1, rendering this pathway kinetically unfavorable [Figure 4A][57]. In contrast, the presence of liquid water activates a Grotthuss-type proton relay mediated by interfacial water molecules, reducing the energy barrier to approximately 47 kJ mol-1 [Figure 4B][57]. This finding highlights the essential role of liquid water: rather than serving merely as a passive solvent, it constructs a low-barrier HBN that dramatically facilitates dynamic proton migration and active-site reconstruction on catalyst surfaces. Cheng et al. further employed random phase approximation (RPA) theory combined with surface-charging methods to analyze the electrochemical environment of water-solid interfaces[58]. Despite perturbation in water adsorption orientations induced by electrode potentials, surface-adsorbed water was shown to maintain cooperative proton transfer via Grotthuss-type HBNs, thereby enabling efficient *CO reduction[58].
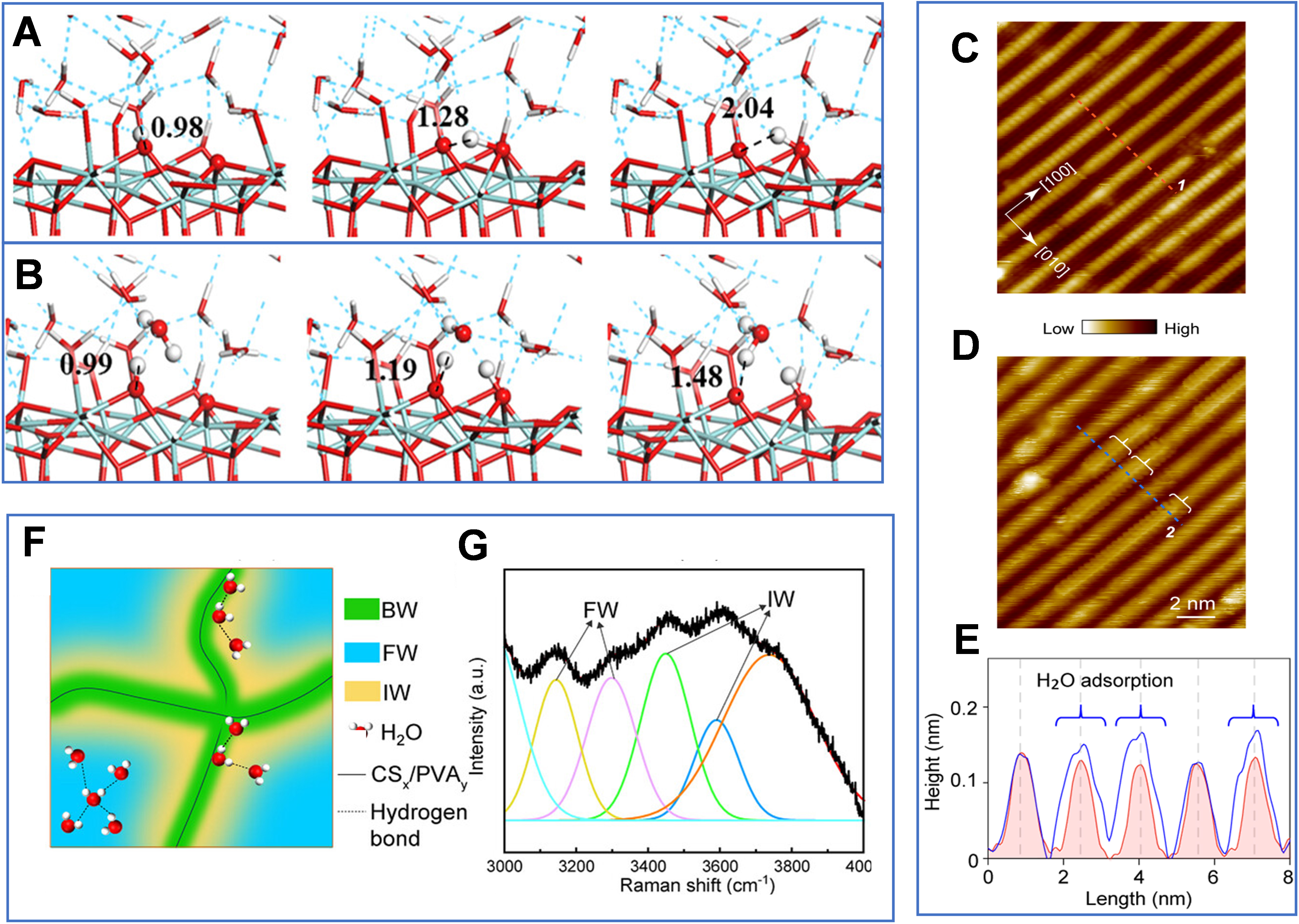
Figure 4. (A) Proton transfer between 2-fold coordinated oxygen (O2c) and 3-fold coordinated oxygen (O3c) sites on the ZrO2(111) surface in condensed water; (B) Proton transfer between O2c and O3c sites on ZrO2(111) mediated by the Grotthuss mechanism. (A and B) Ref.[57] Copyright © 2017 American Chemical Society; (C) Atomically resolved STM images of bare anatase TiO2(001)-(1 × 4) surfaces and water-covered surfaces (acquisition conditions: 1.0 V, 10 pA, 80 K); (D) STM image of the surface after water adsorption (acquisition conditions: 1.2 V, 10 pA, 80 K), where fuzzy stripes represent mobile H2O molecules, and brackets indicate assembled water molecule structures; (E) Line profiles extracted along the marked lines in the STM images of the bare (red) and water-covered (blue) surfaces, and structural diagrams of the H2O-OH monolayer; (C-E) Ref.[60] Copyright © 2022 American Chemical Society; (F) Schematic illustration of three water states and the distribution of water molecules in the hydrogel, including bond water, free water, and interfacial water; (G) Gaussian deconvolution of the O-H stretching band of water confined in the polymer. (F and G) Ref.[72] Copyright © 2025 Wiley-VCH GmbH. BW: Bound water; FW: free water; IW: intermediate water; PVA: poly(vinyl alcohol); STM: scanning tunneling microscopy.
In summary, interactions at the water-solid interface are crucial for overcoming proton-transfer energy barriers. Through chemisorption to surface atoms and HB bridging, interfacial water molecules effectively alleviate spatial constraints imposed by solid lattices[50]. This intimate interfacial coupling preserves long-range Grotthuss channels even under complex electrochemical conditions and establishes the importance of water-assisted mechanisms in lowering activation energies and promoting surface proton flux.
Wetting layers and interfacial configurations
The wetting layer at the water-solid interface, characterized by structures distinct from bulk water, acts as a critical barrier to charge and mass transport[59]. Driven by surface physicochemical properties as well as HB interactions, interfacial water molecules undergo reconstruction to form a “Janus-type” interface with specific dipole orientations[50]. Substrate wettability governs the orientation of water dipoles and induces the assembly of ordered HBNs. Such interfacial ordering not only alters system entropy but also critically determines interfacial charge-transfer efficiency and reaction selectivity.
Ma et al. combined in situ ultraviolet spectroscopy, X-ray photoelectron spectroscopy (XPS), and DFT calculations to investigate water adsorption on anatase TiO2(001)-(1 × 4) surfaces[60]. At sub-monolayer coverage, water molecules exhibit weak interactions with surface 5-fold coordinated Titanium (Ti5c) sites, characterized by physisorption and high lateral mobility. Scanning tunneling microscopy (STM) images and height profiles reveal that as coverage approaches and exceeds one monolayer, water molecules self-assemble into ordered interfacial structures. Additional water molecules shorten O-O distances and construct a complex interfacial HBN, increasing the average adsorption energy to 0.71 eV and stabilizing the wetting layer [Figure 4C-E]. More importantly, this network breaks the spatial isolation of terrace sites and opens cascaded proton and hole transfer pathways, enabling photogenerated holes to efficiently oxidize and dissociate water molecules. Wang et al. revealed that hexagonal boron nitride (h-BN) surfaces exhibit Janus-like behavior driven by a dynamic equilibrium between chemisorbed and physisorbed hydroxide ions[61]. This unique interfacial chemistry induces spontaneous surface charging and markedly strengthens interfacial water structuring, providing a molecular-level explanation for the higher water friction of h-BN compared to graphene[61].
Collectively, these findings demonstrate that the interfacial wetting layer is not a simple extension of bulk water but a structured water phase formed under surface potential constraints. The orientation and HB connectivity of the first water layer thus govern charge distribution, proton transport pathways, and intermediate stability[62,63].
Nanoconfinement and solvation effects on the water-solid interfaces
While surface HBNs capture collective water behavior in two-dimensional interfacial systems, water molecules in practical catalytic and electrochemical energy-storage devices often experience more complex geometric constraints or chemical environments. When molecular degrees of freedom are restricted by nanoscale confinement (e.g., porous materials) or strong ionic fields (e.g., highly concentrated electrolytes), the properties of bulk water are profoundly altered[64-68]. This section examines how confined spaces and solvation structures reshape the physical state and chemical reactivity of water, thereby dictating macroscopic reaction selectivity and stability.
The “shape” of water in nanopores
In nanoporous materials such as zeolites and metal-organic frameworks, pore sizes are often comparable to the dimensions of water clusters (< 2 nm). Such extreme confinement prevents water molecules from sustaining the tetrahedral HBN characteristic of bulk water, forcing them to reorganize into one-dimensional chains, small clusters, or quasi-gaseous monomers with distinct thermodynamic and kinetic properties[69-71].
Zhang et al. constructed a spatially graded hydrophilic-hydrophobic solid-water interface by introducing hydrophobic silica nanoparticles into a porous hydrogel framework[72]. They demonstrated that this engineered solid-water interfacial network can significantly reduce surface water content, regulate the hydrogen-bonding states of water molecules, and effectively decrease the enthalpy of water evaporation[72]. At the solid-water interface, abundant hydrophilic functional groups within the porous hydrogel framework restructure water molecules into three distinct states through interfacial interactions: bound water (BW), free water (FW), and intermediate water (IW) [Figure 4F]. Raman spectroscopic analysis of O-H stretching vibrations [Figure 4G] further confirms that this spatially engineered solid interface effectively weakens the dense hydrogen-bonding network among water molecules and significantly increases the proportion of IW, which exhibits a lower evaporation energy barrier. Consequently, the overall enthalpy of water evaporation is reduced at the microscopic molecular scale, providing a fundamental thermodynamic basis for efficient liquid-vapor phase transitions. Treps et al. used DFT calculations to investigate hydrated ZSM-5 zeolites with different surface orientations[73]. They found that water speciation strongly depends on local topology: at nanopore mouths, water tends to dissociate into cavity-stabilized bridging hydroxyls with strong Brønsted acidity, whereas on outer surfaces it remains molecularly adsorbed, acting as mild Brønsted acid sites whose stability depends on HBNs formed by surface silanol groups[73].
Overall, nanoscale spatial confinement induces water restructuring, providing additional degrees of freedom for regulating reaction processes and transforming water from a background solvent into a key factor influencing reaction pathways and energy barrier distributions[74,75]. Thus, understanding the structural response of water in confined environments is a prerequisite for explaining anomalously enhanced reaction behaviors in water-solid systems.
Solvation reconstruction in battery electrolytes
Unlike confinement-dominated water restructuring in nanopores, solvation reconstruction at water-solid interfaces in electrochemical systems primarily reflects interactions between water molecules and solid surfaces[76,77]. Near interfaces with high surface energy or strong interactions, water molecules couple with surface functional groups, defect sites, and localized charges to form solvation layers with specific orientations and stabilities[78]. These interfacial solvation structures govern not only water adsorption and dissociation but also the stability and migration of protons and reaction intermediates[79].
Wang et al. employed melamine as a multifunctional electrolyte additive to regulate Zn2+ solvation and interfacial interactions, thereby significantly enhancing zinc anode stability[80]. The strongly electronegative groups in melamine exhibit higher binding energies with Zn2+ than water molecules, partially stripping water from the Zn2+ solvation sheath and reconstructing the HB network of the electrolyte, which reduces the HOMO-LUMO gap to accelerate charge transfer and promotes preferential melamine adsorption at the water/zinc interface. Additionally, Joos et al. systematically investigated transport properties across the full concentration range of Li-SCN-H2O electrolytes, revealing that trace water molecules can act as dopants by substituting anions and forming H2O-SCN defects[81]. This mechanism simultaneously increases lithium vacancy concentration and mobility, enhancing ionic conductivity by nearly three orders of magnitude[81].
In conclusion, electrolyte solvation reconstruction arises from the dynamic evolution of chemical environments and intermolecular interactions at water-solid interfaces. Through functional additives or solvent composition tuning, water solvation structures can be precisely engineered at the molecular scale - either by stripping solvation shells to suppress side reactions or by inducing defects to enhance ionic conductivity[80,81]. These insights underscore that water at solid-liquid interfaces is not an inert medium but a highly tunable functional component that directly governs charge transfer and material transformation processes[79].
MACROSCALE FIELD EFFECT ON THE REACTION PROCESS OF WATER AT SOLID INTERFACES
Electric-field-governed solid-water interfaces
The electric field effect at solid-water interfaces serves as a key mechanism governing physicochemical processes. Based on its origin, it can be primarily categorized into two dominant modes: built-in electric fields and externally applied electric fields. The former arises from the intrinsic properties of materials and electrolytes - such as electric double layers, heterojunctions, and surface polarization - and acts as an endogenous driving force for interfacial ion transport, molecular orientation, and electrochemical reactions. The latter is achieved through external application, enabling active and precise interfacial manipulation with dynamically tunable, intelligent characteristics. Here, we systematically elucidate the operating principles of these two electric-field modes, focusing on how they regulate interfacial water structure, ion solvation and transport, and reaction pathways to address scientific and technological challenges in fields such as energy storage, catalysis, and sensing.
Built-in electric fields
The spontaneously formed built-in electric field at the solid-water interface constitutes a central physical factor driving various electrochemical processes. In energy storage systems such as batteries and supercapacitors, this type of field - originating from intrinsic material and electrolyte properties - governs interfacial water structure, ion transport, and interfacial reactions, thereby determining overall device performance. For example, in the field of piezoelectric catalysis, BON materials with asymmetric unit-cell-layer structures and abundant surface hydroxyl groups can eliminate interlayer electric-field shielding and achieve in situ self-polarization, thereby significantly enhancing the material’s piezoelectricity and built-in electric field. This leads to improved mechanical energy conversion efficiency and higher yields in piezocatalytic water splitting[82].
In supercapacitors, Shi et al. constructed a MnO/MnS heterojunction combined with a BCN substrate and utilized the synergistic effect between the heterojunction’s built-in electric field and the BCN matrix to effectively widen the voltage window of an aqueous supercapacitor[83]. This built-in electric field works in concert with the proton-attracting effect of N sites in BCN, markedly enhancing the adsorption and migration of Li+ on the electrode surface. As a result, the electrode surface becomes positively charged during discharge, electrostatically repelling H+ in the electrolyte, thereby suppressing the hydrogen evolution reaction and raising its overpotential. Simultaneously, a reverse electric field forms during charging, accelerating Li+ desorption and further inhibiting the oxygen evolution reaction[83]. Similarly, in zinc-based battery systems, introducing amino acid-based additives into the electrolyte can regulate the solvation structure of Zn2+ and promote the formation of a stable solid-electrolyte interphase layer, thereby suppressing hydrogen evolution and dendrite growth triggered by interfacial water, and improving the corrosion resistance and cycling stability of the zinc anode[84].
Furthermore, introducing a 7,7,8,8-tetracyanoquinodimethane (TCNQ) charge-transfer complex can form a Zn(TCNQ)2 semiconductor interface on the zinc surface. The work-function difference generates a built-in electric field, which homogenizes the electric-field distribution, guides rapid Zn2+ transport, and suppresses water molecule activity, thus alleviating dendrite growth and side reactions on the zinc anode[85]. In summary, by modulating interfacial water structure, ion solvation, and transport behavior, builtin electric fields can effectively suppress side reactions, enhance deposition uniformity, and improve interfacial stability. This provides a crucial physical foundation for addressing performance bottlenecks in a variety of electrochemical devices.
External applied electric fields
In contrast to built-in electric fields, externally applied electric fields provide an effective means for actively and precisely manipulating solid-water interfaces. By applying an electric field through external electrodes, phenomena such as electrowetting, electroosmotic flow, and electric-field-induced phase transitions can be induced[86]. Further studies have shown that tuning the local electric field on Pd/Cu2O catalyst surfaces can guide interfacial water molecules to reorient into an “H-down” configuration, shortening the M-H bond length and promoting water dissociation and active hydrogen generation, thereby enhancing the efficiency and selectivity of the electrochemical nitric oxide reduction reaction for ammonia synthesis[87]. In porous carbon electrode systems, combined model analysis and experimental verification can clarify the relationship between the desolvation state of potassium ions and the critical pore size, while identifying an optimal concentration window for oxygen-containing functional groups. This synergy optimizes both thermodynamic ion adsorption and kinetic diffusion, leading to simultaneous improvements in capacitive performance and rate capability[88].
Furthermore, anchoring quaternary ammonium cations onto hydrophobic covalent organic frameworks (COFs) and modifying copper electrode surfaces can significantly enhance the local electric field at the electrode-electrolyte interface [Figure 5A]. This strengthens CO intermediate adsorption and promotes CO-CO coupling, thereby improving the selectivity and stability of CO2 electroreduction to ethylene. The hydrophobicity and microporous structure of the COFs help regulate the transport of CO2 and H2O, while the immobilized cations modulate K+ migration via the Donnan effect, further intensifying the interfacial electric field[89]. On highly curved RuIr nanocatalyst surfaces, an enhanced local electric field can drive interfacial water into an ordered “O-down” orientation and strengthen the hydrogen-bond network, thereby accelerating the kinetics of the alkaline hydrogen oxidation reaction (HOR) and improving fuel-cell performance[90].
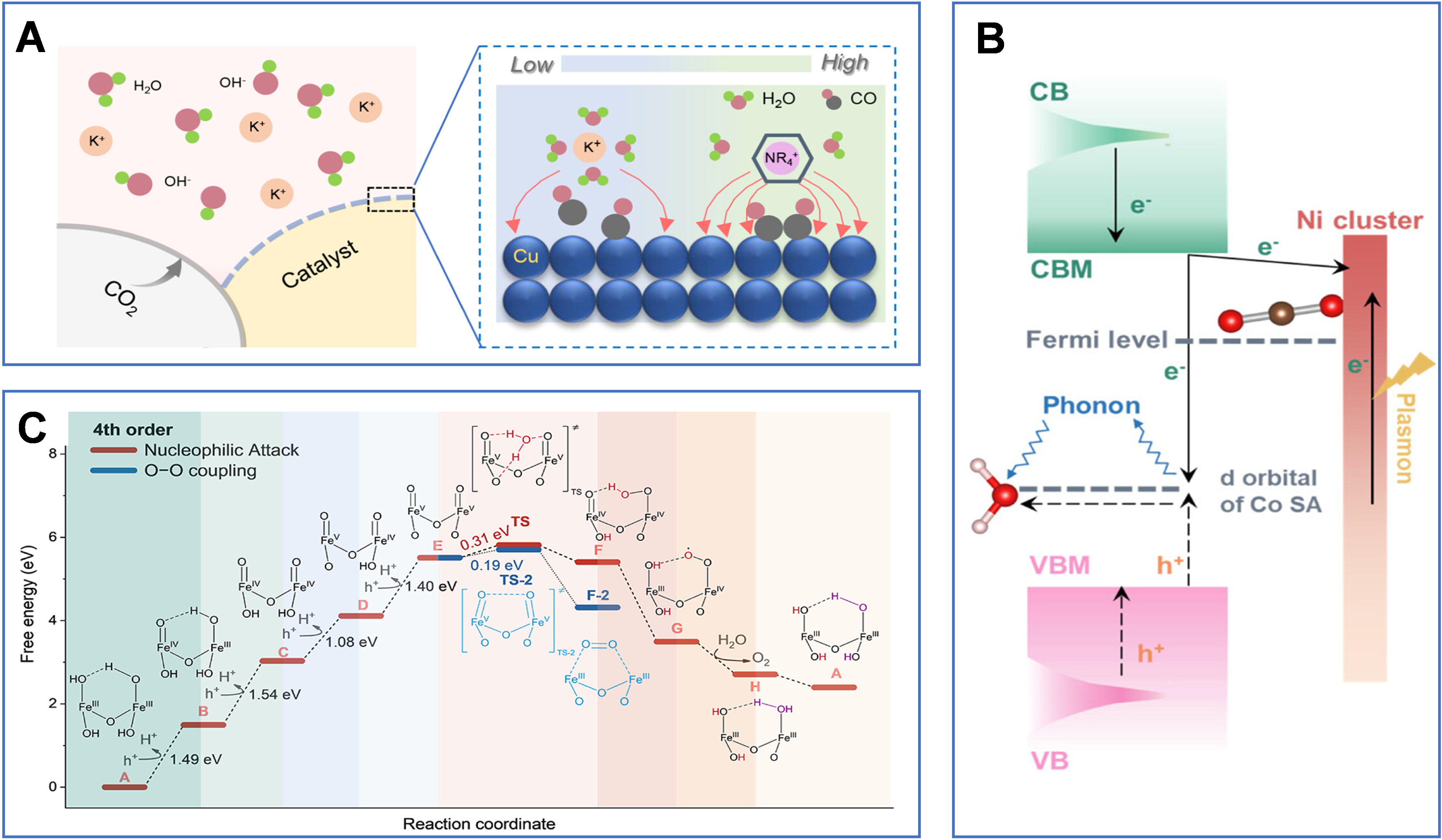
Figure 5. (A) Schematic illustration of the enhanced local electric field at the interface induced by Me-COF modification. Ref.[89] Copyright © 2025 American Chemical Society; (B) Schematic of two pathways for photon-energy conversion. Ref.[92] Copyright © 2024 American Chemical Society; (C) Potential energy surface for water oxidation on the α-Fe2O3 surface. The fourth-order reaction pathway was obtained from DFT calculations. “TS” denotes the transition state for O-O bond formation. All electrochemical steps incorporate a photovoltage of 0.80 V. Ref.[41] Copyright © 2025 American Chemical Society. CB: Conduction band; CBM: conduction band minimum; SA: single atom; VBM: valence band maximum; VB: valence band; TS: transition state; COF: covalent organic framework.
The cases discussed above demonstrate that applied electric fields can exert precise control over water molecular orientation, ion transport, and reaction pathways by modulating the intensity and distribution of local electric fields. This offers an effective strategy for addressing key challenges related to efficiency, selectivity, and stability in electrochemical devices.
Light-field-governed solid-water interfaces
The interaction of light with solid-water interfaces is primarily realized through two physical mechanisms: the photoelectric effect and the photothermal effect. The photoelectric effect converts photon energy into electronic excitation, driving interfacial charge transfer and electrochemical reactions. The photothermal effect transforms light energy into localized heat, altering the interfacial temperature field and mass transfer processes. Acting either synergistically or independently, these two mechanisms provide fundamental pathways for regulating interfacial processes in fields such as solar energy conversion, photocatalysis, and microfluidics.
Photothermal effect
In photothermal catalytic processes at solid-water interfaces, the photothermal effect stems from the conversion of light energy into heat through non-radiative relaxation, generating a non-uniform temperature field at the interface[72]. This, in turn, drives mass transfer and reaction processes by altering physicochemical parameters such as interfacial tension, fluid density, and reactant activation energy. This effect constitutes the physical basis for technologies including solar thermal utilization and photothermal catalysis.
For example, in PtW/TiO2 bimetallic catalysts under photothermal synergistic conditions, acidic hydroxyl groups on W sites can anchor water molecules via hydrogen bonds, transforming the competitive adsorption of O2 and H2O into cooperative activation. This promotes the generation of reactive oxygen species, thereby alleviating the poisoning of volatile-organic-compound oxidation catalysts caused by water molecules in high-humidity, low-temperature environments[91]. Notably, as demonstrated by Chen et al. [Figure 5B], the simultaneous loading of Co single atoms and Ni clusters onto oxygen-vacancy-rich ZrO2 enables strong photothermal coupling under concentrated solar irradiation: the hybridization between the d-orbitals of Co single atoms and the molecular orbitals of H2O forms intermediate impurity states, promoting hole generation and transfer under visible-light excitation and accelerating H2O dissociation[92]. Meanwhile, Ni clusters convert light energy into hot electrons and localized thermal fields via localized surface plasmon resonance, effectively activating CO2 and facilitating C=O bond cleavage. This synergistic mechanism involving spatially separated dual active sites significantly enhances the kinetics and efficiency of solar full-spectrum-driven CO2 reduction with water to CO[92].
These studies illustrate that the photothermal effect, by creating localized thermal fields and modulating interfacial electronic structures, can enhance mass transfer and lower reaction energy barriers, offering an effective strategy for efficiently utilizing solar energy to drive heterogeneous catalytic reactions.
Photoelectric effect
The essence of the photoelectric effect lies in the generation of photogenerated electron-hole pairs and their effective separation and utilization at interfaces. This process enables active control of the electric double-layer structure, carrier injection, and surface reaction kinetics through photovoltage or photocurrent, thereby establishing a coupled “photo-electro-chemical” interplay.
Research has shown that under high-intensity illumination, semiconductor photoanodes (e.g., α-Fe2O3, TiO2, WO3, BiVO4) can accumulate a high density of photogenerated holes at their surfaces[41]. This accumulation drives a shift in the water oxidation reaction mechanism from lower-order kinetics to a nearly barrier-free, fourth-order kinetic pathway [Figure 5C][41]. Both theoretical calculations and experimental results indicate that when four holes accumulate at the active site, adjacent high-valent Fe(V)=O intermediates are formed. This configuration lowers the activation energy for O-O bond formation to about 0.03 eV and increases the reaction rate by more than an order of magnitude relative to conventional first-, second-, or third-order kinetics. The process is facilitated by consecutive proton-coupled electron transfers of surface holes and is further enhanced through hydrogen-bond-mediated activation of interfacial water molecules, leading to a significant improvement in the efficiency and kinetics of photoelectrochemical water oxidation[41].
In terms of heterojunction design, constructing Z-scheme heterojunctions with surface oxygen vacancies (e.g., n-Bi12SiO20/p-Bi2S3) can synergistically enhance visible-light absorption, promote charge separation, and generate reactive oxygen species. This approach collectively improves the photocatalytic removal efficiency of low-concentration nitrogen oxides in high-humidity environments, while also suppressing the formation of toxic by-products[93]. Additionally, introducing N-hydroxymethyl functional groups onto the surface of g-C3N4 can enhance the adsorption and activation of O2, as well as the dehydrogenation kinetics of H2O, while preserving its intrinsic photoelectric properties[94]. Constructing a spin-polarized electric field derived from sulfur vacancies in CdS nanorods enables efficient visible-light-driven overall water splitting for hydrogen production without the need for cocatalysts. By promoting charge separation from the bulk to the surface, this strategy mitigates the severe charge-carrier recombination and high reaction barriers commonly encountered in conventional photocatalysis[95].
In summary, the photoelectric effect - through interface engineering strategies such as defect modulation, heterojunction construction, and surface functionalization - optimizes the generation, separation, and utilization of photogenerated charge carriers, thus offering a key pathway to enhance the efficiency, selectivity, and stability of photocatalytic and photoelectrochemical reactions.
Chemical-field-governed solid-water interfaces
The chemical environment at solid-water interfaces often involves multiple components that collectively shape the interfacial microenvironment through two fundamental modes: competition and synergy. On one hand, competition manifests as the rivalry among different components for limited interfacial sites or reaction pathways, directly regulating interfacial composition, charge, and wettability. On the other hand, synergy, arising from molecular interactions and the ordered assembly of components, can give rise to enhanced functionalities that surpass those of individual constituents. Understanding the dynamic interfacial processes under these two modes is central to tailoring interface performance in applications such as catalysis, corrosion, and separation.
Competition
In multicomponent chemical environments, differing physicochemical properties among species often lead to dynamic competition for adsorption sites, reaction pathways, and interfacial energy at the solid-water interface, thereby significantly influencing interfacial composition, charge distribution, and even macroscopic performance.
Lee et al. employed in situ RAXR to probe ion adsorption at charged solid-water interfaces[96]. Their results show that competition from monovalent cations (Na+/Rb+) induces nonclassical adsorption behavior of the divalent ion Sr2+, driving it from a mixed IS/outer-sphere (OS) configuration toward predominantly OS complexes and significantly weakening its adsorption strength. In particular, Rb+ preferentially occupies IS cavity sites on the surface, forcing Sr2+ to desorb and exist mainly as fully hydrated OS species. To minimize electrostatic repulsion, these OS-coordinated Sr2+ ions further adopt an interleaved spatial arrangement with IS-bound Rb+ at the interface [Figure 6A][96].
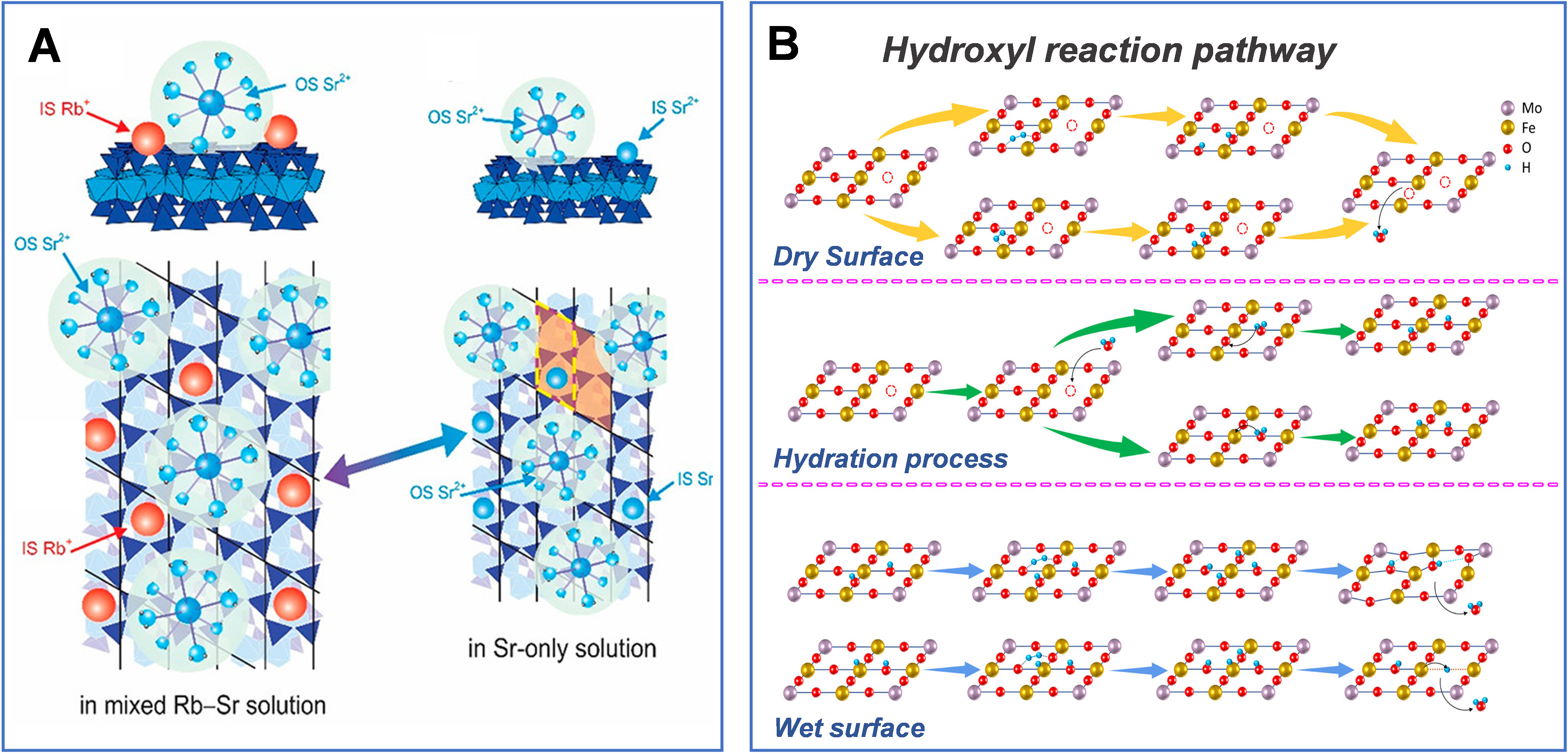
Figure 6. (A) Schematic of the distribution of mixed states of Sr2+ adsorbed on the muscovite mica surface, as inferred from X-ray measurements, and the coexistence of inner-sphere (IS) Rb+ and outer-sphere (OS) Sr2+ adsorption on the mica surface in a solution containing 20 mM SrCl2 and 3 mM RbCl. The lateral distribution of the adsorbates is inferred from X-ray reflectivity data, simulating the strong positional correlations expected at the highly charged mica surface. Ref.[96] Copyright © 2020 American Chemical Society; (B) Hydrogen oxidation reaction pathways on dry (yellow line) and hydrated (blue line) surfaces. The hydration process, transitioning from the dry to the hydrated surface, is indicated by a green line. Ref.[97] Copyright © 2020 American Chemical Society.
On the other hand, Zhou et al. combined in situ spectroscopy and theoretical calculations to elucidate that carbonate anions (CO32-) and their derived radicals (CO3•-) can form stable hydration layers with water molecules via HBs, promoting the structuring of the interfacial water network[98]. This ordered water layer not only reduces the energy barrier for proton migration - accelerating the Volmer step of the hydrogen evolution reaction - but also enables CO3•⁻ to serve as an additional carbon source that can be reduced to formate or CO. Consequently, a dynamic competitive relationship is established between CO2 reduction and hydrogen evolution, ultimately leading to the suppression of CO2 reduction at high cathodic potentials. This finding provides a molecular-level understanding of how anions modulate competing reaction pathways through the structuring of hydration layers[97,98]. Similarly, on γ-Al2O3 surfaces, gas molecules such as H2O, SO2, and CO2 compete with gaseous arsenic pollutants (As2O3) for limited surface sites - a mechanism that explains, at the microscopic level, fluctuations in arsenic adsorption efficiency in multicomponent flue gas[99]. In the low-temperature selective catalytic reduction (SCR) of NOx with NH3 over Mn-Cu-Al layered oxide catalysts, water molecules compete with the reactants NH3 and NOx for surface active sites and promote the formation of less reactive hydroxylated nitrate intermediates, thereby significantly suppressing low-temperature denitration activity[100].
In summary, the competitive adsorption and reaction of multiple components at the interface constitute a central factor in determining the interfacial chemical state and, ultimately, the functional output. Managing this dynamic interplay allows for the optimization of the interfacial microenvironment, thereby enabling the fine-tuning of catalytic selectivity, reaction efficiency, and material stability.
Synergy
In contrast to competition, the coexistence of multiple chemical species at the interface can give rise to synergy through intermolecular interactions, sequential adsorption, or the formation of composite interfacial phases, leading to enhanced functionality.
At the surface of perovskite anodes (e.g., Sr2Fe1.5Mo0.5O6-δ[97,101]) in solid oxide fuel cells, water molecules in humid hydrogen atmospheres can synergistically enhance the kinetics of HOR by lowering the surface oxygen vacancy concentration and generating surface hydroxyl species, thereby improving cell performance. Specifically, Qi et al. combined electrochemical relaxation and in situ spectroscopic analysis to reveal that water molecules promote the HOR on Sr2Fe1.5Mo0.5O6 (SFM)-based perovskites through two synergistic mechanisms [Figure 6B][97]. On one hand, the introduction of H2O increases the surface oxygen chemical potential and reduces the near-surface oxygen nonstoichiometry (δ) and the electron chemical potential, thereby lowering the energy barrier of the rate-limiting step involving “H2O adsorption and surface oxygen vacancy formation” in the HOR. On the other hand, surface hydroxyl and hydride species formed via water dissociation can further stabilize HOR intermediates by strengthening the interaction between surface hydrogen species and lattice oxygen or oxygen vacancies, thereby accelerating surface reaction kinetics. This work elucidates, from both energetic and structural perspectives, the “co-catalytic” synergistic mechanism of water molecules at perovskite anode surfaces[98].
Similarly, on the surface of arsenopyrite (FeS2), the pre-adsorption of water molecules enhances the binding of oxygen and significantly lowers the energy barrier for arsenic oxidation, thereby clarifying the microscopic mechanism by which water vapor synergistically promotes the formation and release of toxic arsenic oxides during combustion[102]. Additionally, the use of layered rare-earth oxycarbonates (Ln2O2CO3) enables efficient and reversible exchange between surface hydroxyl groups (from H2O dissociation) and carbonate species (from CO2 adsorption). This “molecular exchange” mechanism allows for the timely removal of the gaseous product CO2, thereby preventing its conversion into stable carbonates that would otherwise poison active sites. In this way, it addresses the critical issue of catalyst deactivation due to carbon deposition in the water-gas shift reaction[103].
These cases demonstrate that through the rational design of interfaces to harness synergy among components, it is possible to construct functional interfaces with high performance and stability, providing a significant paradigm for the design of advanced materials and catalytic systems.
INTERFACIAL BEHAVIOR OF WATER AT SOLID SURFACES
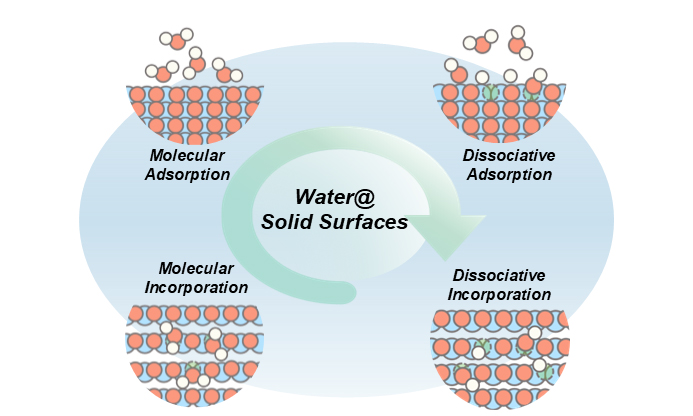
Building on the multiscale framework outlined above and illustrated in Figure 7, this review systematically analyzes the complex behavior of water at solid-water interfaces. To distill the common physicochemical principles underlying these behaviors, we return to the fundamental chemical nature of water-solid interactions - specifically, whether water molecules undergo dissociation. By further considering the sites of interaction, we classify interfacial processes into two broad categories encompassing four core modes: molecular adsorption, dissociative adsorption, molecular incorporation, and dissociative absorption. This classification reveals, at a foundational level, the mechanisms of charge-carrier generation and transport at interfaces. Molecular-scale interactions primarily modulate ion mobility by altering the local environment or structure, as demonstrated in various systems (e.g., superacidic sulfated zirconia[104] and Nafion ionomers[105]). In contrast, dissociative interactions directly introduce new proton or hydroxide ion carriers - a key route to achieving high ionic conductivity, particularly proton conduction, as observed on nanoporous CeO2 surfaces[106]. Molecular incorporation into the crystal bulk can modulate bulk carrier concentration and mobility, as exemplified in NaOH systems[107]. Dissociative absorption within the bulk fills lattice defects and introduces intrinsic proton defects, representing a key strategy for designing high-temperature fast ion conductors such as perovskite-type oxides[108,109].
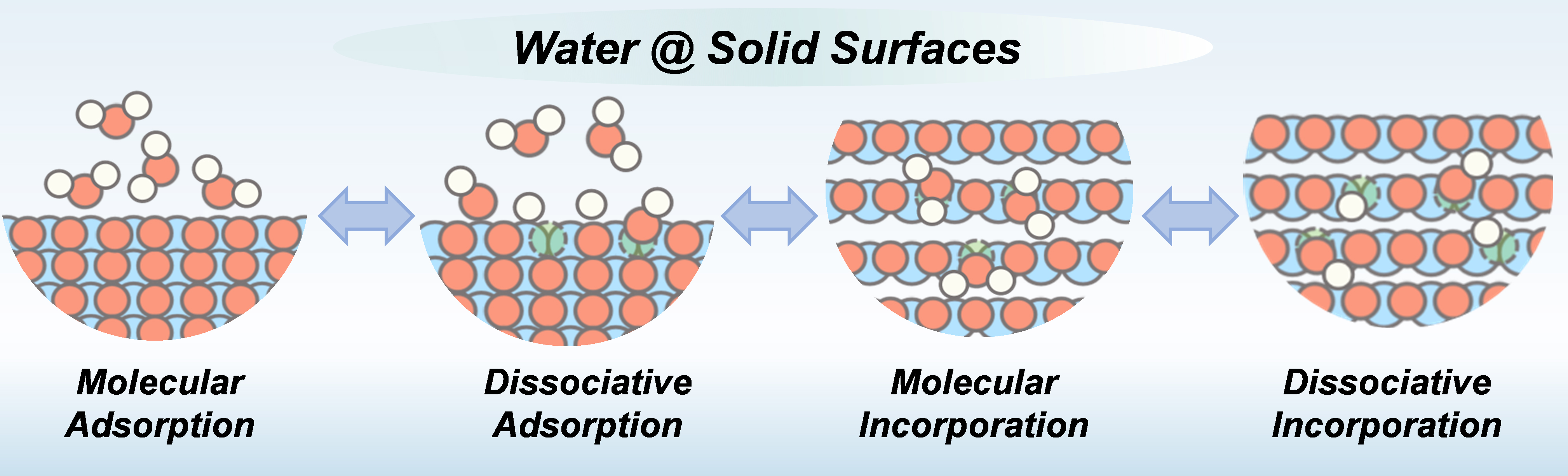
Figure 7. Summary of governing principles at the solid-water interface.
CONCLUSION AND OUTLOOK
In this review, we summarize theoretical simulation studies of interfacial ion behavior from atomic to macroscopic scales. At the atomic and molecular levels, we discuss the static binding properties, electronic structures, and dynamic solvation mechanisms between ions and surface sites. At the mesoscale, we focus on the collective diffusion, competitive adsorption, and interfacial transport of ions in HBNs and nanoconfined spaces. At the macroscopic scale, we explain how these microscopic processes control colloidal stability, pollutant migration, reactive transport, and system performance through electric field control, photocatalysis, and molecular competition. Furthermore, we discuss the potential states of water at solid surfaces and classify them into two broad categories, including four core modes of molecular adsorption, dissociative adsorption, molecular incorporation, and dissociative absorption. This review helps solve the problems of chemical complexity and multiscale effects, providing a systematic theoretical basis and methodological support for predicting and controlling interfacial ion behavior in real environmental and engineering applications.
Research on solid-water interfaces is entering a new phase, shifting from phenomenological description toward mechanistic prediction and functional design. Future advances will depend on the deep integration and intelligent development of multiscale simulation methods. In particular, the creation of interpretable, adaptive machine learning potentials capable of bridging spatial and temporal scales will be essential to accurately capture complex interfacial reaction pathways and kinetics, building on recent successes in simulating facet-dependent water structures and dynamics. Concurrently, progress in ultra-high spatiotemporal-resolution in situ characterization will open a window for observing transient interfacial processes, providing critical validation for theoretical models, as demonstrated in recent studies of interfacial water structure in CO2 electroreduction[97]. Given the complexity of real processes, where multiple components and physical fields are coupled, constructing integrated multiphysics models that incorporate chemical, electric, optical, and flow fields will form a key bridge from microscopic mechanisms to macroscopic performance prediction - as illustrated by recent advances in electric field regulation[83,87] and photoelectric integration[92]. Building on this foundation, the rational design of interfacial materials for applications such as environmental remediation, energy conversion and storage, nanoionic devices, and photo-/electrocatalysis will become attainable, ultimately translating scientific understanding into technological solutions. This progression urgently requires deep interdisciplinary collaboration and the development of open-source software, shared databases, and coordinated experimental platforms to accelerate the formation of a fully integrated innovation cycle linking theory, simulation, experiment, and application.
DECLARATIONS
Authors’ contributions
Writing - original draft: Chen, H.; Wang, S.
Figures and Material support: Guo, Z.; Lu, J.
Administrative support, manuscript revision and editing: Wang, Y.; He, H.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This research was funded by the National Natural Science Foundation of China (22278401, 22425806, and 22522817), the U35 (New Teacher Start-Up Fund) Project of Renmin University of China (25XNKJ13), and the Research Project of Longzihu New Energy Laboratory (LZH2023001-04). It was also supported by the Public Computing Cloud, Renmin University of China.
Conflicts of interest
Wang, Y.; He, H. are Associate Editors of the journal Iontronics. Wang, Y.; He, H. were not involved in any stage of the editorial process, notably including reviewer selection, manuscript handling, or decision making. The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Bañuelos, J. L.; Borguet, E.; Brown, G. E.; et al. Oxide- and silicate-water interfaces and their roles in technology and the environment. Chem. Rev. 2023, 123, 6413-544.
2. Langmuir, I. The constitution and fundamental properties of solids and liquids. J. Frankl. Inst. 1917, 183, 102-5.
3. Hayes, K. F.; Roe, A. L.; Brown, G. E.; Hodgson, K. O.; Leckie, J. O.; Parks, G. A. In situ X-ray absorption study of surface complexes: selenium oxyanions on α-FeOOH. Science 1987, 238, 783-6.
4. Brown, G. E.; Henrich, V. E.; Casey, W. H.; et al. Metal oxide surfaces and their interactions with aqueous solutions and microbial organisms. Chem. Rev. 1998, 99, 77-174.
5. Hiemstra, T.; Van Riemsdijk, W. A Surface structural approach to ion adsorption: the charge distribution (CD) model. J. Colloid. Interface. Sci. 1996, 179, 488-508.
6. Sverjensky, D. A. Zero-point-of-charge prediction from crystal chemistry and solvation theory. Geochim. Cosmochim. Acta. 1994, 58, 3123-9.
7. Lee, S. S.; Fenter, P.; Nagy, K. L.; Sturchio, N. C. Real-time observation of cation exchange kinetics and dynamics at the muscovite-water interface. Nat. Commun. 2017, 8, 15826.
8. Fumagalli, L.; Esfandiar, A.; Fabregas, R.; et al. Anomalously low dielectric constant of confined water. Science 2018, 360, 1339-42.
9. Eng, P. J.; Trainor, T. P. Brown Jr., G. E.; et al. Structure of the hydrated α-Al2O3(0001) surface. Science 2000, 288, 1029-33.
10. Trainor, T. P.; Chaka, A. M.; Eng, P. J.; et al. Structure and reactivity of the hydrated hematite (0001) surface. Surf. Sci. 2004, 573, 204-24.
11. Zhang, C.; Hutter, J.; Sprik, M. Coupling of surface chemistry and electric double layer at TiO2 electrochemical interfaces. J. Phys. Chem. Lett. 2019, 10, 3871-6.
12. Kumar, N.; Kent, P. R. C.; Bandura, A. V.; et al. Faster proton transfer dynamics of water on SnO2 compared to TiO2. The. Journal. of. Chemical. Physics. 2011, 134, 044706.
13. Rother, G.; Stack, A. G.; Gautam, S.; Liu, T.; Cole, D. R.; Busch, A. Water uptake by silica nanopores: impacts of surface hydrophilicity and pore size. J. Phys. Chem. C. 2020, 124, 15188-94.
14. Ilgen, A. G.; Kabengi, N.; Leung, K.; Ilani-Kashkouli, P.; Knight, A. W.; Loera, L. Defining silica-water interfacial chemistry under nanoconfinement using lanthanides. Environ. Sci:. Nano. 2021, 8, 432-43.
15. Wang, R.; Dellostritto, M.; Remsing, R. C.; Carnevale, V.; Klein, M. L.; Borguet, E. sodium halide adsorption and water structure at the α-alumina(0001)/water interface. J. Phys. Chem. C. 2019, 123, 15618-28.
16. Yang, J.; Xie, T.; Mei, Y.; et al. High-efficiency V-Mediated Bi2MoO6 photocatalyst for PMS activation: modulation of energy band structure and enhancement of surface reaction. Appl. Catal. B. Environ. 2023, 339, 123149.
17. Su, R.; Liu, Z.; Qiu, J.; et al. Photoexcited Hole‐Enabled Synthesis of Surface High‐Valent Cobalt‐Oxo Species with Water as the Oxygen Atom Source for Water Purification. Angew. Chem. Int. Ed. 2025, 64, e202507085.
18. Peng, R.; Ren, Y.; Si, Y.; et al. Strong photothermal tandem catalysis for CO2 reduction to C2H4 boosted by Zr-O-W interfacial H2O dissociation. ACS. Catal. 2024, 15, 1-13.
19. Jianchun, S.; Zhoulin, L.; Qiang, W.; et al. Glycine-mediated hydrogen bond network reconstruction and interfacial engineering for ultrahigh-voltage aqueous magnesium air batteries. Chem. Eng. J. 2025, 520, 166111.
20. Zhang, Z.; Wen, L.; Jiang, L. Nanofluidics for osmotic energy conversion. Nat. Rev. Mater. 2021, 6, 622-39.
21. Ge, C.; Wang, M.; Zhou, Y.; et al. Ion transport-triggered rapid flexible hydrovoltaic sensing. Nat. Commun. 2025, 16, 8110.
22. Cuba-chiem, L. T.; Huynh, L.; Ralston, J.; Beattie, D. A. In situ particle film ATR FTIR Spectroscopy of carboxymethyl cellulose adsorption on talc: binding mechanism, pH effects, and adsorption kinetics. Langmuir 2008, 24, 8036-44.
23. Knight, A. W.; Kalugin, N. G.; Coker, E.; Ilgen, A. G. Water properties under nano-scale confinement. Sci. Rep. 2019, 9, 8246.
24. Knight, A. W.; Ilani-kashkouli, P.; Harvey, J. A.; et al. Interfacial reactions of Cu(ii) adsorption and hydrolysis driven by nano-scale confinement. Environ. Sci:. Nano. 2020, 7, 68-80.
25. Kubicki, J. D.; Tunega, D.; Kraemer, S. A density functional theory investigation of oxalate and Fe(II) adsorption onto the (010) goethite surface with implications for ligand- and reduction-promoted dissolution. Chem. Geol. 2017, 464, 14-22.
26. Gittus, O. R.; Von Rudorff, G. F.; Rosso, K. M.; Blumberger, J. Acidity constants of the hematite-liquid water interface from ab initio molecular dynamics. J. Phys. Chem. Lett. 2018, 9, 5574-82.
27. Holmboe, M.; Bourg, I. C. Molecular dynamics simulations of water and sodium diffusion in smectite interlayer nanopores as a function of pore size and temperature. J. Phys. Chem. C. 2013, 118, 1001-13.
28. Kerisit, S.; Rosso, K. M. Kinetic Monte Carlo model of charge transport in hematite (α-Fe2O3). J. Chem. Phys. 2007, 127, 124706.
29. Neumann, A.; Wu, L.; Li, W.; et al. Atom exchange between aqueous Fe(II) and structural Fe in clay minerals. Environ. Sci. Technol. 2015, 49, 2786-95.
30. Barry, E.; Burns, R.; Chen, W.; et al. Advanced materials for energy-water systems: the central role of water/solid interfaces in adsorption, reactivity, and transport. Chem. Rev. 2021, 121, 9450-501.
31. Bi, F.; Meng, Q.; Zhang, Y.; et al. Engineering triple O-Ti-O vacancy associates for efficient water-activation catalysis. Nat. Commun. 2025, 16, 851.
32. Chen, J.; Meng, Y.; Xie, B.; Ni, Z.; Xia, S. Construction of ultrathin BiVO4 nanosheets with bismuth-oxygen dual vacancies for photocatalytic nitrogen reduction. Chin. J. Catal. 2025, 78, 265-78.
33. Raizada, P.; Soni, V.; Kumar, A.; et al. Surface defect engineering of metal oxides photocatalyst for energy application and water treatment. J. Materiomics. 2021, 7, 388-418.
34. Chang, Y.; Zhou, W.; Chen, Y.; et al. Oxygen vacancies in ultrathin BiOCl nanosheets induced Pt for enhanced methanol oxidation. Process. Saf. Environ. Prot. 2025, 196, 106866.
35. Wang, Y.; Zhang, Y.; Zhu, X.; Liu, Y.; Wu, Z. Fluorine-induced oxygen vacancies on TiO2 nanosheets for photocatalytic indoor VOCs degradation. Appl. Catal. B. Environ. 2022, 316, 121610.
36. Chen, X.; He, G.; Li, Y.; et al. Identification of a facile pathway for dioxymethylene conversion to formate catalyzed by surface hydroxyl on TiO2-based catalyst. ACS. Catal. 2020, 10, 9706-15.
37. Huang, J.; Yang, S.; Jiang, S.; Sun, C.; Song, S. Entropy-increasing single-atom photocatalysts strengthening the polarization field for boosting H2O overall splitting into H2. ACS. Catal. 2022, 12, 14708-16.
38. Chen, W.; Zheng, W.; Cao, J.; et al. Atomic insights into robust Pt-PdO interfacial site-boosted hydrogen generation. ACS. Catal. 2020, 10, 11417-29.
39. Li, H.; Shang, J.; Zhu, H.; Yang, Z.; Ai, Z.; Zhang, L. Oxygen vacancy structure associated photocatalytic water oxidation of BiOCl. ACS. Catal. 2016, 6, 8276-85.
40. Albanese, E.; Di Valentin, C.; Pacchioni, G. H2O Adsorption on WO3 and WOx (001) surfaces. ACS. Appl. Mater. Interfaces. 2017, 9, 23212-21.
41. Liu, S.; Dang, K.; Wu, L.; Bai, S.; Zhang, Y.; Zhao, J. Nearly barrierless four-hole water oxidation catalysis on semiconductor photoanodes with high density of accumulated surface holes. J. Am. Chem. Soc. 2025, 147, 4520-30.
42. Lin, L.; Zhou, W.; Gao, R.; et al. Low-temperature hydrogen production from water and methanol using Pt/α-MoC catalysts. Nature 2017, 544, 80-3.
43. Niu, S.; Wang, J.; Wu, Y.; et al. Constructing a built-in electric field to accelerate water dissociation for efficient alkaline hydrogen evolution. ACS. Appl. Mater. Interfaces. 2024, 16, 31480-8.
44. Qiao, B.; Wang, A.; Yang, X.; et al. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nature. Chem. 2011, 3, 634-41.
45. Wang, A.; Li, J.; Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018, 2, 65-81.
46. Su, B.; Kong, Y.; Wang, S.; et al. Hydroxyl-bonded Ru on metallic TiN surface catalyzing CO2 reduction with H2O by infrared light. J. Am. Chem. Soc. 2023, 145, 27415-23.
47. Michaelides, A.; Morgenstern, K. Ice nanoclusters at hydrophobic metal surfaces. Nat. Mater. 2007, 6, 597-601.
49. Fujitani, T.; Nakamura, I.; Takahashi, A. H2O dissociation at the perimeter interface between gold nanoparticles and TiO2 is crucial for oxidation of CO. ACS. Catal. 2020, 10, 2517-21.
50. Björneholm, O.; Hansen, M. H.; Hodgson, A.; et al. Water at interfaces. Chem. Rev. 2016, 116, 7698-726.
51. Khatib, R.; Backus, E. H. G.; Bonn, M.; Perez-haro, M.; Gaigeot, M.; Sulpizi, M. Water orientation and hydrogen-bond structure at the fluorite/water interface. Sci. Rep. 2016, 6, 24287.
52. Shi, B.; Pang, X.; Li, S.; et al. Short hydrogen-bond network confined on COF surfaces enables ultrahigh proton conductivity. Nat. Commun. 2022, 13, 6666.
53. Maiyelvaganan, K.; Kamalakannan, S.; Shanmugan, S.; Prakash, M.; Coudert, F.; Hochlaf, M. Identification of a Grotthuss proton hopping mechanism at protonated polyhedral oligomeric silsesquioxane (POSS) - water interface. J. Colloid. Interface. Sci. 2022, 605, 701-9.
54. Zhao, R.; Wang, Q.; Yao, Y.; et al. Pd single atoms guided proton transfer along an interfacial hydrogen bond network for efficient electrochemical hydrogenation. Sci. Adv. 2025, 11, eadu1602.
55. Groß, A.; Sakong, S. Ab initio simulations of water/metal interfaces. Chem. Rev. 2022, 122, 10746-76.
56. Farnesi Camellone, M.; Negreiros Ribeiro, F.; Szabová, L.; Tateyama, Y.; Fabris, S. Catalytic proton dynamics at the water/solid interface of ceria-supported Pt clusters. J. Am. Chem. Soc. 2016, 138, 11560-7.
57. Cai, Q.; Lopez-ruiz, J. A.; Cooper, A. R.; Wang, J.; Albrecht, K. O.; Mei, D. Aqueous-phase acetic acid ketonization over monoclinic zirconia. ACS. Catal. 2017, 8, 488-502.
58. Cheng, D.; Wei, Z.; Sautet, P. Elucidating the proton source for CO2 electro-reduction on Cu(100) using many-body perturbation theory. J. Am. Chem. Soc. 2025, 147, 10954-65.
59. Baidoun, R.; Liu, G.; Kim, D. Recent advances in the role of interfacial liquids in electrochemical reactions. Nanoscale 2024, 16, 5903-25.
60. Ma, X.; Shi, Y.; Liu, J.; et al. Hydrogen-bond network promotes water splitting on the TiO2 surface. J. Am. Chem. Soc. 2022, 144, 13565-73.
61. Wang, Y.; Luo, H.; Advincula, X. R.; et al. Spontaneous surface charging and janus nature of the hexagonal boron nitride-water interface. J. Am. Chem. Soc. 2025, 147, 30107-16.
62. Wang, Y.; Zheng, S.; Yang, W.; et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 2021, 600, 81-5.
63. Shi, G.; Lu, T.; Zhang, L. Understanding the interfacial water structure in electrocatalysis. Natl. Sci. Rev. 2024, 11, nwae241.
64. Melnik, S.; Ryzhov, A.; Kiselev, A.; et al. Confinement-controlled water engenders unusually high electrochemical capacitance. J. Phys. Chem. Lett. 2023, 14, 6572-6.
65. Augustyn, V.; Gogotsi, Y. 2D materials with nanoconfined fluids for electrochemical energy storage. Joule 2017, 1, 443-52.
66. Liang, T.; Hou, R.; Dou, Q.; Zhang, H.; Yan, X. The applications of water-in-salt electrolytes in electrochemical energy storage devices. Adv. Funct. Mater. 2020, 31, 2006749.
67. Wang, C.; Bai, P.; Siepmann, J. I.; Clark, A. E. Deconstructing hydrogen-bond networks in confined nanoporous materials: implications for alcohol-water separation. J. Phys. Chem. C. 2014, 118, 19723-32.
68. Li, C.; Chen, M.; Liu, S.; et al. Unconventional interfacial water structure of highly concentrated aqueous electrolytes at negative electrode polarizations. Nat. Commun. 2022, 13, 5330.
69. Furukawa, H.; Gándara, F.; Zhang, Y.; et al. Water adsorption in porous metal-organic frameworks and related materials. J. Am. Chem. Soc. 2014, 136, 4369-81.
70. Rieth, A. J.; Hunter, K. M.; Dincă, M.; Paesani, F. Hydrogen bonding structure of confined water templated by a metal-organic framework with open metal sites. Nat. Commun. 2019, 10, 4771.
71. Coudert, F.; Boutin, A.; Fuchs, A. H. Open questions on water confined in nanoporous materials. Commun. Chem. 2021, 4, 106.
72. Zhang, J.; Chen, S.; Wang, Y.; Guo, W.; Li, H.; He, H. Spatially Janus-like solar evaporator with high evaporation rate and enhanced salt processing capacity. Adv. Funct. Mater. 2025, 36, e26145.
73. Treps, L.; Gomez, A.; De Bruin, T.; Chizallet, C. Environment, stability and acidity of external surface sites of silicalite-1 and ZSM-5 micro and nano slabs, sheets, and crystals. ACS. Catal. 2020, 10, 3297-312.
74. Cored, J.; Wang, M.; Akter, N.; et al. Water formation reaction under interfacial confinement: Al0.25Si0.75O2 on O-Ru(0001). Nanomaterials 2022, 12, 183.
75. Wang, M.; Zhou, C.; Akter, N.; Tysoe, W. T.; Boscoboinik, J. A.; Lu, D. Mechanism of the accelerated water formation reaction under interfacial confinement. ACS. Catal. 2020, 10, 6119-28.
76. Bampoulis, P.; Sotthewes, K.; Dollekamp, E.; Poelsema, B. Water confined in two-dimensions: fundamentals and applications. Surf. Sci. Rep. 2018, 73, 233-64.
77. Shimizu, T. K.; Maier, S.; Verdaguer, A.; Velasco-Velez, J.; Salmeron, M. Water at surfaces and interfaces: from molecules to ice and bulk liquid. Prog. Surf. Sci. 2018, 93, 87-107.
78. Tang, Z.; Dong, Z.; Yuan, L.; Li, B.; Zhu, Y. Unlocking the potential: key roles of interfacial water in electrocatalysis. EES. Catal. 2025, 3, 943-71.
79. Dai, Y.; Zhang, C.; Zhang, X.; et al. Interfacial energy storage in aqueous zinc-ion batteries. Energy. Environ. Sci. 2025, 18, 9018-30.
80. Wang, X.; Peng, H.; Sun, K.; et al. Melamine induced co-regulation of solvation structure and interface engineering to achieve dendrite-free Zn-ion hybrid capacitors. Energy. Storage. Mater. 2024, 66, 103208.
81. Joos, M.; Conrad, M.; Münchinger, A.; et al. Lithium ion transport in water-containing Li(SCN) over a wide compositional range: from water doping to hydration. Solid. State. Ion. 2023, 394, 116130.
82. Wang, C.; Tu, S.; Chen, F.; Ma, T.; Huang, H. Asymmetric single-unit-cell layer enriching polar inherent hydroxyls eliminates interlayer electric field shielding effect and in situ self-polarize for piezocatalytic water splitting. Adv. Mater. 2025, 37, 2505592.
83. Shi, D.; Yang, M.; Zhang, B.; et al. BCN‐assisted built-in electric field in heterostructure: an innovative path for broadening the voltage window of aqueous supercapacitor. Adv. Funct. Mater. 2021, 32, 2108843.
84. Wang, H.; Wang, K.; Liang, B.; et al. Solvation modification and interfacial chemistry regulation via amphoteric amino acids for long-cycle zinc batteries. Adv. Energy. Mater. 2024, 14, 2402123.
85. Xiong, P.; Lin, C.; Wei, Y.; et al. Charge-transfer complex-based artificial layers for stable and efficient Zn metal anodes. ACS. Energy. Lett. 2023, 8, 2718-27.
86. Jiang, Z.; Bazianos, P. P.; Yan, Z.; Rappe, A. M. Mechanism of water dissociation with an electric field and a graphene oxide catalyst in a bipolar membrane. ACS. Catal. 2023, 13, 7079-86.
87. Bai, C.; Wang, J.; Fan, S.; Li, X.; Liu, S. Modulating the reorientation of interfacial water to promote electrochemical NO reduction to NH3 on palladium-copper catalysts. ACS. Catal. 2025, 15, 9442-51.
88. Xia, Y.; Dong, W.; Yang, S.; et al. Decoding potassium ion desolvation states for enhanced electric double-layer capacitance in actual porous carbon. Small 2025, 21, e10058.
89. Qian, Z.; Liu, Y.; Lin, Z.; et al. Hydrophobic cation-immobilized covalent organic frameworks enable selective and stable electrosynthesis of ethylene from CO2. J. Am. Chem. Soc. 2025, 147, 21877-84.
90. Li, L.; Guo, H.; Lv, F.; et al. Curvature-engineered interfacial hydrogen-bond networks driving proton-coupled electron transfer boosts hydrogen oxidation in alkaline fuel cells. Nat. Commun. 2025, 16, 11461.
91. Wang, X.; Zhou, X.; Yan, Z.; et al. Constructing a robust water-resistant PtW/TiO2 catalyst for synergistic photothermocatalytic VOCs elimination. Appl. Catal. B. Environ. 2026, 385, 126249.
92. Chen, J.; Ren, Y.; Fu, Y.; et al. Integration of Co single atoms and Ni clusters on defect-rich ZrO2 for strong photothermal coupling boosts photocatalytic CO2 reduction. ACS. Nano. 2024, 18, 13035-48.
93. Chang, F.; Yang, C.; Wang, X.; et al. Mechanical ball-milling preparation and superior photocatalytic NO elimination of Z-scheme Bi12SiO20-based heterojunctions with surface oxygen vacancies. J. Clean. Prod. 2022, 380, 135167.
94. Liu, B.; Du, J.; Ke, G.; et al. Boosting O2 reduction and H2O dehydrogenation kinetics: surface N‐hydroxymethylation of g‐C3N4 photocatalysts for the efficient production of H2O2. Adv. Funct. Mater. 2021, 32, 2111125.
95. He, J.; Hu, L.; Shao, C.; Jiang, S.; Sun, C.; Song, S. Photocatalytic H2O overall splitting into H2 bubbles by single atomic sulfur vacancy CdS with spin polarization electric field. ACS. Nano. 2021, 15, 18006-13.
96. Lee, S. S.; Park, C.; Sturchio, N. C.; Fenter, P. Nonclassical behavior in competitive ion adsorption at a charged solid-water interface. J. Phys. Chem. Lett. 2020, 11, 4029-35.
97. Qi, H.; Lee, Y.; Yang, T.; et al. Positive Effects of H2O on the hydrogen oxidation reaction on Sr2Fe1.5Mo0.5O6-δ-based perovskite anodes for solid oxide fuel cells. ACS. Catal. 2020, 10, 5567-78.
98. Zhou, Y.; Ibáñez-Alé, E.; López, N.; Roldan Cuenya, B.; Kley, C. S. Carbonate anions and radicals induce interfacial water ordering in CO2 electroreduction on gold. Nature. Chem. 2025, 18, 473-81.
99. Hu, P.; Weng, Q.; Li, D.; Lv, T.; Wang, S.; Zhuo, Y. Effects of O2, SO2, H2O and CO2 on As2O3 adsorption by γ-Al2O3 based on DFT analysis. J. Hazard. Mater. 2021, 403, 123866.
100. Gui, R.; Zhang, C.; Gao, Y.; Wang, Q.; Efstathiou, A. M. Unravelling the multiple effects of H2O on the NH3-SCR over Mn2Cu1Al1Ox-LDO by transient kinetics and in situ DRIFTS. Appl. Catal. B. Environ. 2025, 361, 124611.
101. Farias, M. B.; Araújo, A. J.; Holz, L. I.; et al. Sr2Fe1·5Mo0·5O6-δ-based cathodes for solid oxide fuel cells: Microstructural considerations and composite formation with Pr-doped ceria. Int. J. Hydrogen. Energy. 2024, 61, 1305-16.
102. Zou, C.; Wang, C.; Chen, L.; Zhang, Y.; Xing, J.; Anthony, E. J. The effect of H2O on formation mechanism of arsenic oxide during arsenopyrite oxidation: experimental and theoretical analysis. Chem. Eng. J. 2020, 392, 123648.
103. Zhou, L.; Li, S.; Ma, C.; et al. Promoting molecular exchange on rare-earth oxycarbonate surfaces to catalyze the water-gas shift reaction. J. Am. Chem. Soc. 2023, 145, 2252-63.
104. Hara, S. Proton conductivity of superacidic sulfated zirconia. Solid. State. Ionics. 2004, 168, 111-6.
105. Morris, D. R.; Sun, X. Water‐sorption and transport properties of Nafion 117 H. J. Appl. Polym. Sci. 2003, 50, 1445-52.
106. Sun, X.; Vøllestad, E.; Rørvik, P. M.; et al. Surface protonic conductivity in chemisorbed water in porous nanoscopic CeO2. Appl. Surf. Sci. 2023, 611, 155590.
107. Spaeth, M.; Kreuer, K.; Maier, J.; Cramer, C. Giant haven ratio for proton transport in sodium hydroxide. J. Solid. State. Chem. 1999, 148, 169-77.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0









Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.