


Gut microbial metabolites: emerging roles in vascular aging and implications for disease
 0
0 Abstract
Population aging is a critical global health challenge that drives a significant increase in the burden of aging-related chronic diseases. Vascular aging, defined as the progressive structural and functional degeneration of blood vessels, is a pivotal manifestation of systemic aging and a major contributor to cardiovascular morbidity and mortality. Recent advances have highlighted the gut microbiota and its metabolites as crucial modulators of host physiology and disease. This review synthesizes current knowledge on the "gut-vascular axis", focusing on the mechanistic roles of key gut-derived metabolites such as short-chain fatty acids, bile acids, and phenylalanine-derived metabolites in vascular aging. We provide a detailed analysis of how specific microbial metabolites influence these processes, discuss their implicated roles in aging-related vascular diseases, particularly atherosclerosis and aortic aneurysms, and illustrate their potential as both pathogenic drivers and therapeutic targets.
Keywords
INTRODUCTION
Longer life expectancy and lower fertility rates have made population aging a central challenge for the global healthcare system. According to the United Nations World Population Prospects 2024[1], the global population aged 65 and older may reach 2.2 billion by the late 2070s, and individuals aged 80 and older are projected to reach 265 million by the mid-2030s. This demographic transition has markedly increased the prevalence of aging-related chronic diseases[2]. Vascular aging, the progressive structural and functional degeneration of blood vessels as we age, is a critical aspect of the aging process[3]. Given that aging is both an irreversible biological process and a significant independent risk factor for multiple chronic diseases, it is essential to elucidate the pathogenesis of underlying vascular aging to enable accurate prognosis and to identify potential therapeutic targets.
With the rapid advancement of high-throughput multi-omics technologies and genetic engineering techniques, a large number of studies on the correlations and even causal relationships between the gut microbiota, its metabolites and various diseases have emerged continuously, underscoring the pivotal role of the intestinal microecology in maintaining the health of the body and the occurrence and development of diseases[4]. Accumulating evidence indicates that gut dysbiosis is closely linked to various diseases such as type 2 diabetes mellitus (T2DM), cardiovascular diseases (CVDs), autoimmune diseases, cancers, and neuropsychiatric disorders. Thus, the "gut-vascular axis" has emerged as a novel concept for understanding vascular aging and associated CVDs[5,6].
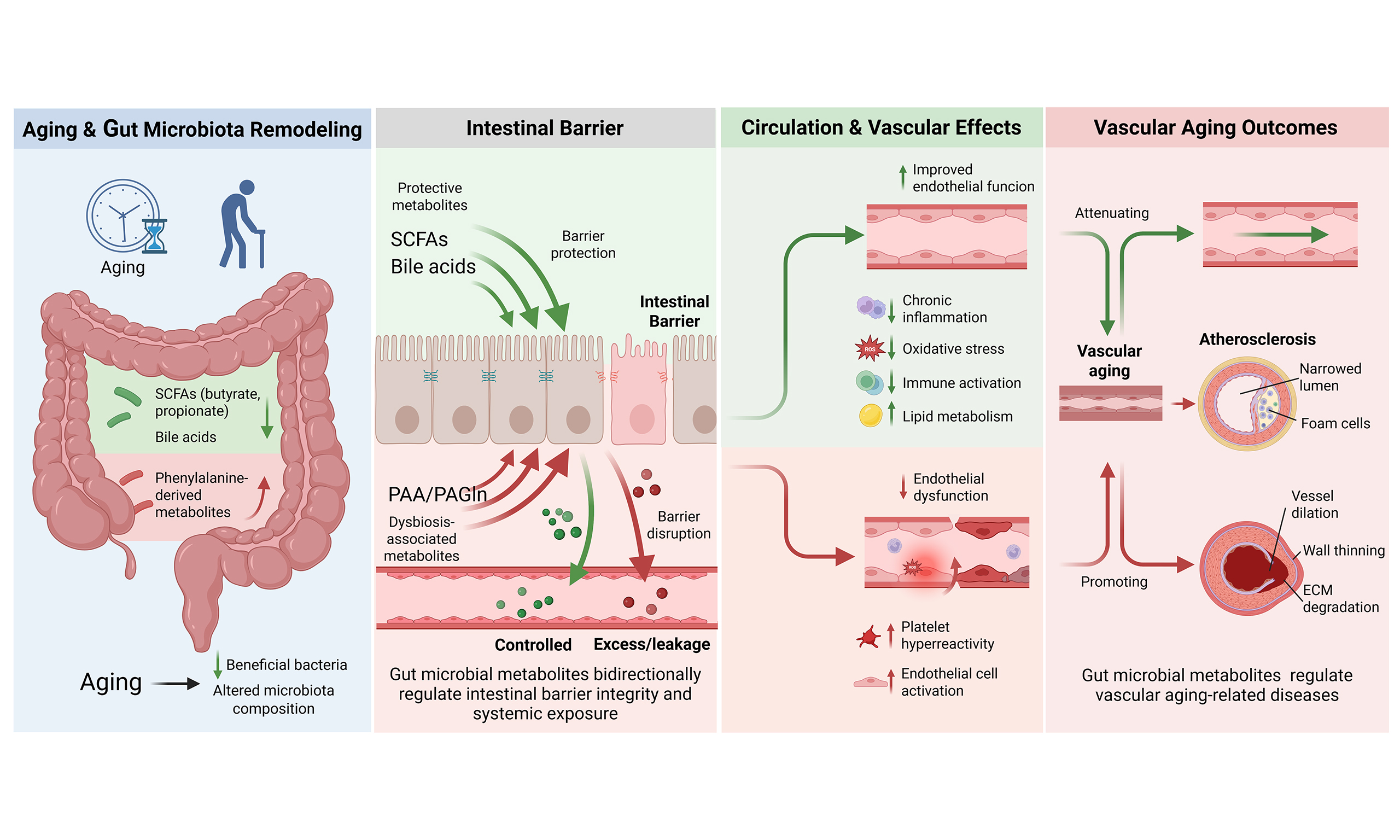
As the aging process progresses, the composition and function of the gut microbiota undergo significant changes due to dietary and environmental factors such as low dietary fiber intake, long-term residential care, antibiotic exposure, and drug use[7] [Figure 1]. These changes are characterized by a decline in species diversity, increased inter-individual variation, and a reduction in beneficial bacteria[8]. In terms of microbiota structure, the elderly have a higher proportion of Bacteroidetes while young adults have higher proportions of Firmicutes and Actinomycetota, so the decreased ratio of Firmicutes/Bacteroidetes is a potential indicator of aging[9,10]. In healthy elderly individuals, beneficial bacteria such as Bifidobacterium, Lactobacillus, Faecalibacterium, and Akkermansia are relatively enriched[7,8,11]. As for microbiota metabolites, studies have shown that under controlled conditions, the abundance of microbiota related to short-chain fatty acids (SCFAs) and their metabolic pathways decreases with age. Acetate supplementation or high-fiber diets improve vascular endothelial function and reduce aortic stiffness in old, but not young, mice[12]. Butyrate and propionate also have the effect of improving endothelial function[12,13]. During aging, bile acid (BA) metabolism is also reshaped: the activity of cholesterol 7α-hydroxylase (cytochrome P450 7A1, CYP7A1) decreases in the old mice, which leads to a reduction in BA synthesis. At the same time, the microbiota with bile salt hydrolase (BSH) activity decreases, resulting in a reduction in the generation of unconjugated BAs and secondary BAs, weakening their anti-inflammatory and anti-infective effects[10]. Reduced fecal BAs can promote cholesterol accumulation in the liver and increase serum LDL levels[10]. The genes related to the generation of phenylacetic acid (PAA) in the gut microbiota of the elderly are more abundant, contributing to increased phenylacetylglutamine (PAGln) levels[14]. PAGln has been shown to promote platelet activation and aggregation, induce endothelial dysfunction (ED), and drive cellular senescence, accelerating vascular aging[15]. Increasing evidence indicates that the changes in microbiota metabolites such as SCFAs, BAs, and PAA/PAGln caused by aging, through affecting intestinal barrier function, metabolite circulation, and inflammatory responses, jointly participate in the occurrence and development of vascular diseases[5,6]. Arterial stiffness is one of key phenotypes of vascular aging. The bibliometric profile of arterial stiffness metabolomics shows that molecules involved in gut microbiota metabolic reactions such as fatty acids, BAs, and amino acids constitute an important research direction in this field[16]. Focusing on these key metabolic pathways and their representative metabolites will help to further clarify the impact of gut microbiota on vascular aging.
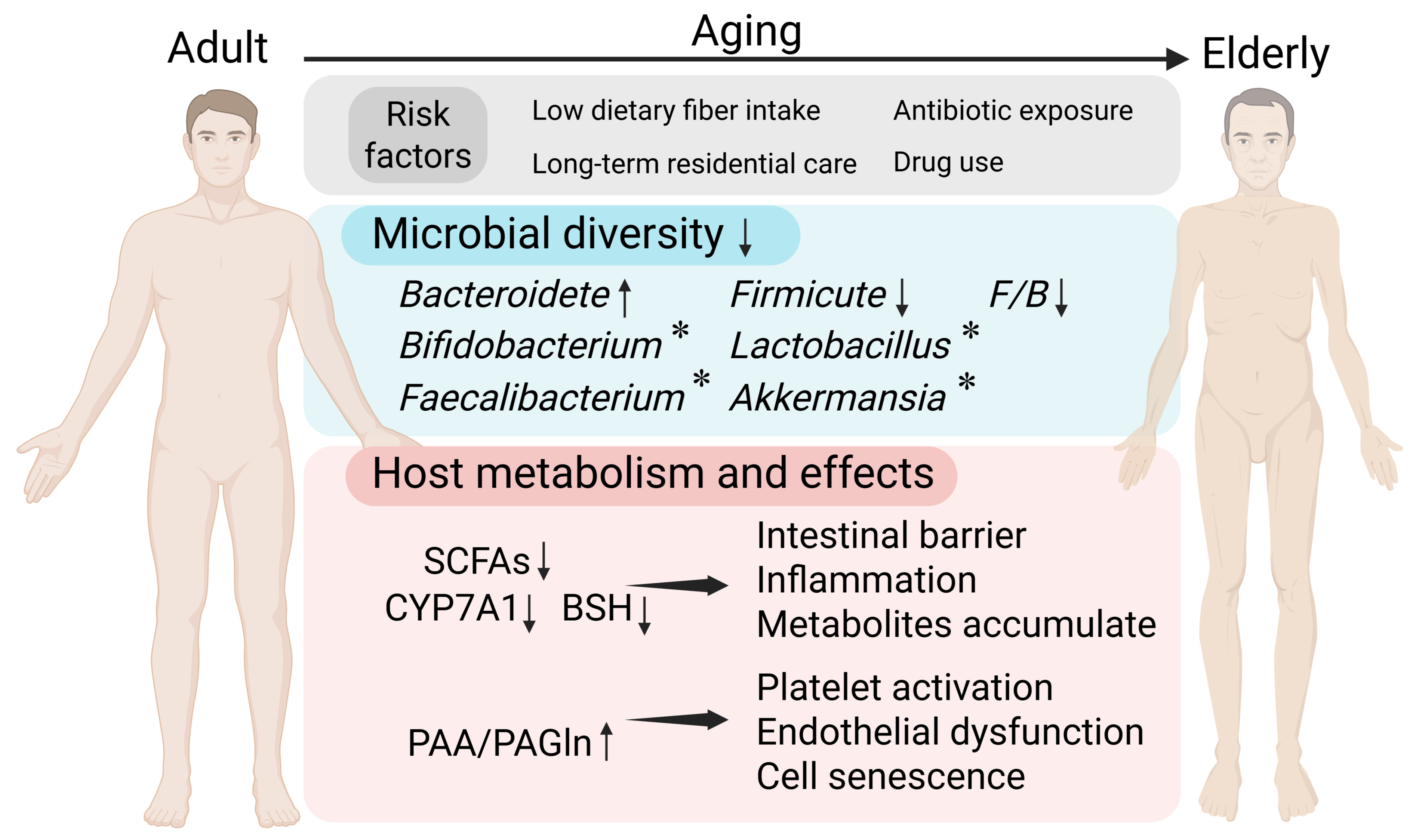
Figure 1. Changes in microorganisms and their metabolites with host aging. Under exposure to multiple risk factors, microbial diversity declines in aging individuals, and the alterations in bacterial genera vary across individuals. Meanwhile, the levels of short-chain fatty acids (SCFAs) and the activity of bile acid (BA)-related metabolic enzymes decrease, whereas phenylalanine-derived metabolite levels increase, eliciting multiple biological effects. (Created in BioRender. Raitasi, E. (2026) https://BioRender.com/dbhuh23). F: Firmicute;
This review summarizes current knowledge on the gut microbiota and vascular aging. First, we define vascular aging and its key features; second, we detail its mechanisms, focusing on ED, vascular smooth muscle cell (VSMC) phenotypic switching, and immune cell infiltration; third, we review how major gut-derived metabolites such as SCFAs, BAs, and phenylalanine-derived metabolites influence vascular aging through different molecular pathways; fourth, we discuss potential causal associations between gut microbial metabolites and vascular aging-related diseases, based on the latest evidence; finally, we critically analyze the limitations of current research in this field and suggest future directions.
VASCULAR AGING
The 17th-century physician Thomas Sydenham, who proposed that "man's arteries age with man", linked vascular aging to organismal aging[17]. The modern "vascular aging theory", developed four centuries later, supports this view and identifies vascular aging as a key factor of multi-organ and systemic aging[18-20]. Understanding the mechanisms of vascular aging is important for improving aging-associated CVDs.
Vascular aging is a process in which the structure and function of arteries gradually deteriorate over time due to long-term exposure to various risk factors such as genetics and the environment. Eventually, this leads to damage in various tissues and organs such as the kidneys, brain, and heart[21]. The basic pathological and physiological changes include the accumulation of pro-inflammatory cytokines, chemokines, and reactive oxygen species in the vascular microenvironment, which causes local cellular phenotypic alterations. Cellular senescence coexists with phenotypic switching, characterized by irreversible proliferation arrest in some cells and enhanced proliferation, migration, and secretory capacity in others[17]. At the same time, the extracellular matrix (ECM) undergoes remodeling, with collagen accumulation, elastin breakdown, and vascular wall calcification. This results in thickening of the arterial media, decreased vascular compliance, and changes in the lumen structure. These changes ultimately manifest as typical characteristics such as ED, arterial stiffness, and elevated blood pressure[17]. Some scholars have referred to it as the "proinflammatory arterial stiffness syndrome"[22].
ED and arterial stiffness are the key phenotypes of vascular aging[23]. The former reflects early functional alterations, while the latter represents cumulative structural remodeling of the arterial wall[24]. Arterial stiffness, defined as a reduction in arterial compliance resulting from both functional and structural changes, precedes the onset of cardiovascular disease and is increasingly recognized as an independent predictor of adverse clinical outcomes[25]. Due to its good reproducibility and standardization, arterial stiffness has emerged as a clinically feasible biomarker that can reflect biological vascular aging beyond chronological age[26]. Currently, commonly used assessment indicators include carotid-femoral pulse wave velocity (c-f PWV), augmentation Index, cardio-ankle vascular index, which are significant in predicting the occurrence and prognosis of cardiovascular events[21]. The impaired functions of endothelial cells are the consequence and inducer of vascular aging[27]. Therefore, ED can not only be characterized as a pathological state but also an important driving factor of arterial stiffness. ED is characterized by reduced nitric oxide bioavailability, increased oxidative stress and inflammation, impaired barrier integrity, and endothelial cell senescence[28]. Clinically, flow-mediated dilation, reactive hyperemia index and certain circulating biomarkers such as NO, adhesion molecules, and coagulation factors are often used to assess endothelial function[28].
Since ED and ECM degradation are the initial components of the pathophysiology of arterial stiffness, and the endothelial cells, as a dynamic regulatory group, deeply participate in the maintenance of vascular microenvironment homeostasis and pathological progression[24], this article regards them together with VSMC phenotypic switching and immune cell infiltration as important mechanisms of vascular aging. These processes are not only the result of the long-term effects of multiple upstream risk factors, but also play a core mediating role in the continuous deterioration of vascular structure and function[24] [Figure 2].
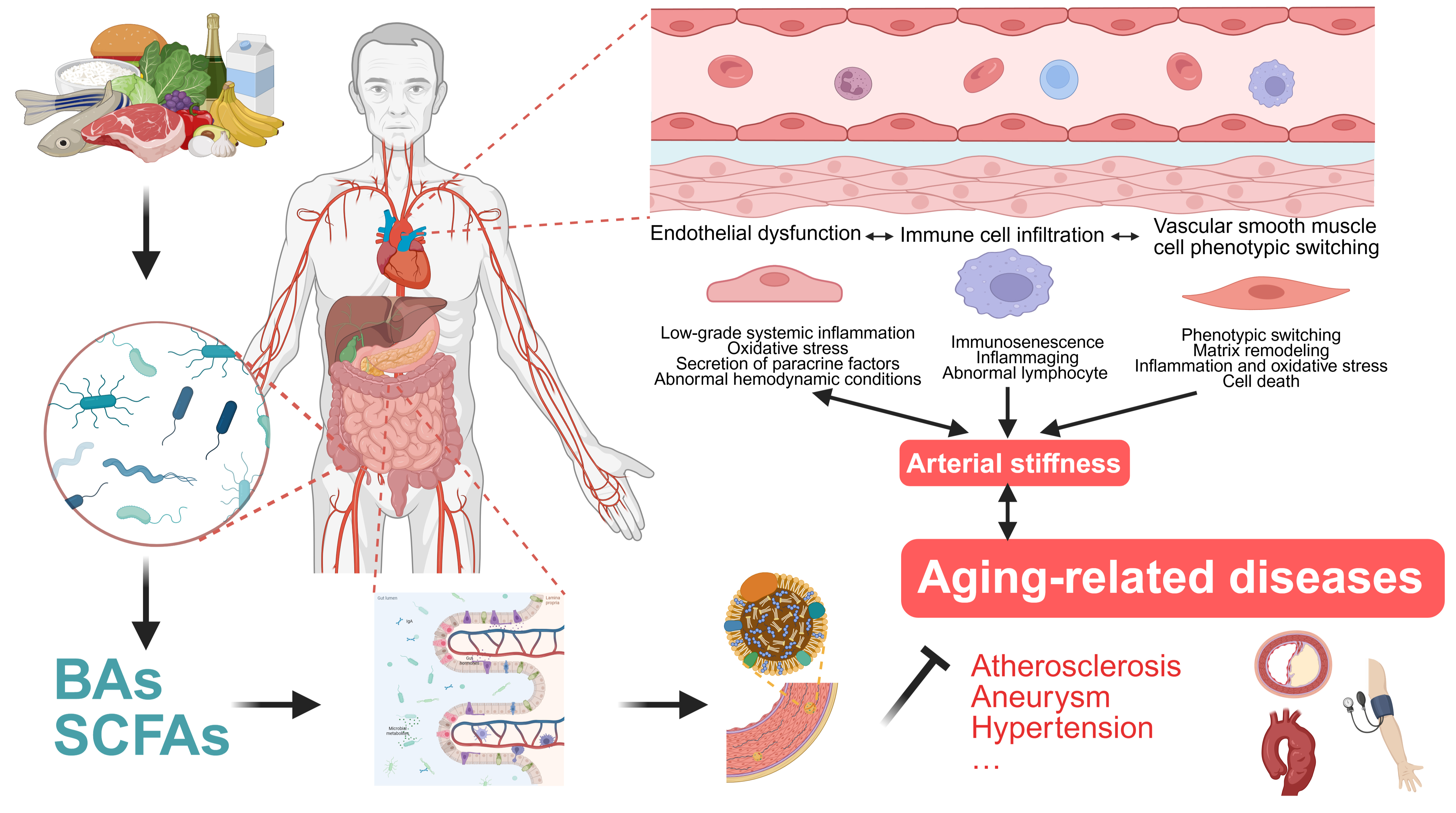
Figure 2. Gut microbial metabolites regulate vascular aging and aging-related diseases. Vascular aging is a complex, multifactorial process driven by interacting mechanisms, among which endothelial dysfunction (ED), vascular smooth muscle cell (VSMC) phenotypic switching, and immune cell infiltration play central roles in its initiation and progression (Created in BioRender. Raitasi, E. (2026) https://BioRender.com/u120b5i). Bas: Bile acids; SCFAs: short-chain fatty acids.
Endothelial dysfunction
Many risk factors drive chronic low-grade systemic inflammation, characterized by immune cell activation and elevated pro-inflammatory cytokines. This promotes reactive oxygen species (ROS) production and upregulates adhesion molecules including intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). The resulting inflammation leads to ED, ECM remodeling, and vascular fibrosis[29]. Low-grade systemic inflammation is coupled with sustained sympathetic nervous system activation[30], which triggers renin angiotensin aldosterone system (RAAS)-mediated upregulation of mineralocorticoid receptor signaling and NADPH oxidase (NOX) activity. These pathways generate excess ROS that impair endothelial integrity[31]. Arterial stiffness can also cause adverse hemodynamic effects. Increased pulsatile amplitude and pressure wave fluctuations increase local shear stress, suppressing NO availability. In contrast, healthy vessels allow endothelial NO synthase activation during normal blood flow, which supports endothelium integrity[32]. Endothelial cells (ECs) also secrete paracrine factors including transforming growth factor-β (TGF-β) and platelet-derived growth factor (PDGF), directly regulating the phenotypic states and functions of VSMCs and fibroblasts. Dysfunction in ECs amplifies pathological signals in the vessel wall, especially with aging[33].
Studies in aged mice and humans show that ECs increase the expression of methyltransferase-like 14 (METTL14), a methyltransferase. This raises N6 methyladenosine (m6A) modification of Toll-like receptor 4 (TLR4) mRNA, stabilizing TLR4 and activating the TLR4-myeloid differentiation primary response 88-nuclear factor-κB (TLR4-MyD88-NF-κB) inflammatory pathway. The pathway fosters a senescence-associated secretory phenotype (SASP), OS, and inflammation, which drive endothelial aging[34]. Single-cell and bulk transcriptomic analyses show lower LRRC8A in aged ECs. This limits AMP-activated protein kinase-sirtuin 1 (AMPK-SIRT1) pathway activity, which normally blocks p53-driven senescence. Reduced LRRC8A also weakens forkhead box O3 (FOXO3)-dependent antioxidant defenses, lowering the endogenous protective capacity of the blood vessel against aging[35].
VSMC phenotypic switching
Phenotypic switching is a typical feature of VSMCs during aging. Under physiological conditions, contractile VSMCs can temporarily transform into synthetic phenotypes in response to minor injuries, enhancing their proliferation and migration and secreting proteases, such as matrix metalloproteinases (MMPs), to participate in ECM remodeling and tissue repair[36]. However, when damage factors such as hypertension, hyperglycemia, and hyperlipidemia accumulate over time and exceed the cells' self-repair capacity, VSMCs shift from a self-restricted synthetic state to an SASP that is maladaptive. SASP-type VSMCs highly express cytoskeletal proteins, integrins, focal adhesion molecules, MMPs, and ECM components including type I and III collagens, which increase ECM stiffness and cause arterial wall thickening and reduced compliance[37]. Under the effects of a high calcium and phosphorus environment, inflammatory and oxidative stress, some senescent VSMCs upregulate osteogenic-related genes such as Runt-related transcription factor 2 (Runx2), bone morphogenetic protein 2 (BMP2), and alkaline phosphatase (ALP), transforming into osteogenic-like phenotypes and driving the process of vascular calcification[38]. Additionally, immune-inflammatory signals, such as complement C3, NOD-like receptor protein 3 (NLRP3) inflammasomes, and extracellular vesicles derived from inflammatory cells, can directly induce VSMCs to switch to inflammatory and synthetic phenotypes. This shift enhances the expression of inflammatory factors and MMPs, ultimately accelerating vascular aging[39-41].
Recent advances have elucidated that multiple pathways encompassing energy metabolism, epigenetic modifications, and cell death programs coordinate VSMC senescence. VSMC uptake of plasma-free fatty acids activates p38/c-Jun N-terminal kinase (p38/JNK) signaling and reduces the expression of ubiquitin ligase C-terminus of heat shock cognate 70 (HSC70)-interacting protein (CHIP), stabilizing of the deacetylase sirtuin 6 (SIRT6), which is no longer protected from proteasomal degradation by CHIP. The loss of SIRT6 binding leads to excessive acetylation of telomeric histones and telomere damage, resulting in telomere integrity loss, which triggers VSMC senescence and eventually causes atherosclerosis (AS)[42]. Similarly, regarding SIRT6 stability, extracellular vesicles enriched with the exerkine fibronectin type-III domain-containing protein 5 (FNDC5) mediate the protective effect of physical activity on blood vessels by increasing SIRT6 stability in VSMCs in a DnaJ/Hsp40 chaperone-dependent manner[43]. During aging, selective suppression of activating transcription factor 3 (ATF3) in VSMCs impairs their capacity to activate autophagy-related 7 (ATG7), which disrupts the ATF3-ATG7 positive feedback loop. This impairment results in autophagosomal depletion and attenuated defenses against senescence and phenotypic switching, culminating in accelerated vascular aging and atherosclerotic progression[44]. In senescent VSMCs, Ras-driven overexpression of tumor necrosis factor receptor-associated protein 1 (TRAP1) amplifies aerobic glycolysis, leading to lactate accumulation, which inhibits histone deacetylase 3 (HDAC3) and promotes histone H4 lysine 12 lactylation (H4K12la). Enrichment of H4K12la at promoters of SASP genes drives senescencerelated transcriptional programs and exacerbates VSMC senescence[45]. In caspase 3gasdermin E (GSDME)-knockout mice, abdominal aortic aneurysm (AAA) lesions show decreased infiltration of proinflammatory macrophages, an increased proportion of tissueresident and immunoregulatory macrophages, and blockade of neutrophil conversion to Cxcr2+ immunosenescent phenotypes. These findings suggest that GSDME-driven pyroptosis coordinates VSMC senescence and coupled SASP activation via immune cell-mediated pathways, accelerating aortic aneurysm pathogenesis[46]. Pro-ferroptotic signaling disrupts peroxisome proliferator-activated receptor-γ (PPAR-γ) nuclear-cytoplasmic shuttling and facilitates ferritinophagy centered on nuclear receptor coactivator 4 (NCOA4)-ferritin protein complexes. This process precipitates vascular NAD+ depletion, cellular senescence, vascular remodeling, and loss of vascular compliance[47].
Immune cell infiltration
With aging, the immune system experiences immunosenescence and inflammaging. Immunosenescence refers to reduced adaptive immune function that includes impaired antimicrobial defense and immune surveillance, along with sustained innate immune activation due to chronic antigen exposure. Inflammaging is a systemic, low-grade, chronic inflammatory state driven by immunosenescence and occurs independently of obvious pathogenic triggers. These processes mutually promote each other and form a vicious cycle, leading to various age-related pathological phenomena: immunosenescence increases the secretion of pro-inflammatory cytokines and SASP factors, while persistent chronic inflammation leads to dysfunction and exhaustion of adaptive immune cells[48]. At the vascular level, aging-related immune remodeling accelerates vascular aging through multiple mechanisms. Senescent macrophages are a rich source of pro-inflammatory mediators and MMPs, which facilitate ECM degradation and elastic fiber fragmentation, and they play a pivotal role in the progression of vascular remodeling and arterial stiffening[49]. Senescent T and B cells progressively acquire a terminal differentiation phenotype. This oligoclonal expansion leads to the loss of immune diversity and the accumulation of cytotoxic subpopulations. Furthermore, the generation of autoantibodies and aberrant signaling interactions with antigen-presenting cells can intensify damage to the vascular microenvironment[50,51].
Through these multifaceted mechanisms, vascular aging has undergone a fundamental shift from a passive, timedependent senescence to an active, multifactorial pathological process. Understanding and studying related vascular diseases from the perspective of vascular aging has become an important direction in cardiovascular research.
GUT MICROBIAL METABOLITES
The human gut microbiota is a complex ecosystem of bacteria, viruses, fungi, parasites, and archaea that has over 3.3 million genes, roughly 150 times more than the human genome. Through continuous co-evolution with the host, this microbial community forms a dynamic superorganism that is constantly reshaped by time and environmental factors[52]. This complex microbial ecosystem accounts for approximately
Gut microbiota dysbiosis, which includes compositional and functional disturbances caused by dietary changes, prolonged or inappropriate use of drugs (particularly antibiotics and nonsteroidal anti‑inflammatory drugs), enteric infections, chronic systemic diseases, and psychosocial stress, exerts a critical pathogenic role in the development of chronic disease[5]. Lifelong continuous antigenic stimulation by gut microbiota can induce senescence in intestinal cells and form a mutually reinforcing vicious cycle with it. This process is accompanied by an increase in pro-inflammatory factor release and a decrease in protective factor secretion, disrupting intestinal homeostasis[57]. Such alterations are thought to underlie the increase in intestinal permeability. Impairment of the gut barrier, in turn, represents a critical contributor to systemic immune activation and the progression of organismal aging[58].
In vascular aging research, the focus has shifted from integral microbiota-level research toward granular, mechanistic investigation of individual microbial taxa and their metabolic outputs. The first large-scale human cohort study to directly associate the gut microbiota with arterial stiffness reported that gut microbial diversity and the relative abundance of several specific taxa were significantly and independently inversely correlated with measures of arterial stiffness in women. Only a small fraction of these associations was mediated by metabolic syndrome components and circulating inflammatory markers, implying that modulation of the gut microbiota could be a promising strategy to attenuate vascular aging[59]. Mice that have been exposed to Western diet for a long time have dysregulated gut microbiota, along with increased arterial stiffness, ED. Notably, the treatment with broad-spectrum antibiotics has been successfully shown to ameliorate these vascular alterations[60]. A Similar study showed that transplanting the gut microbiota of obese individuals into germ-free mice can also cause vascular dysfunction in mice[61]. These studies reveal the potential pathogenic role of gut microbiota imbalance in human blood vessels. Another research that improved the method for identifying bacterial communities found an association between several taxa and arterial stiffness. Moreover, different taxa may have different effects on central and peripheral arterial stiffness[62]. Paralleling the mentioned discovery, Luo and colleagues, starting from a single bacterial species, Flavonifractor plautii and its primary metabolic product cis-aconitic acid, demonstrated how the bacterium-metabolite axis reduces arterial stiffness, advancing the field from population-level to species- and molecule-level analysis[63]. Overall, although technical and experimental limitations often preclude a systematic, end-to-end tracing of the “microbiota-metabolite-vascular aging” axis, focusing on gut-derived metabolites offers a dual advantage: it facilitates identification of candidate molecules suitable for drug targeting or replacement therapies, and it provides crucial leads for back‑tracking to the producing taxa, enabling mechanistic dissection of how specific microbial communities influence vascular aging. This review examines major metabolite categories that are currently the focus of research, namely SCFAs, BAs, and phenylalanine-derived metabolites, and discusses their roles and mechanisms in vascular aging and related diseases.
Short-chain fatty acids
SCFAs are saturated fatty acids with carbon chain lengths of 2-6, primarily produced by colonic bacterial fermentation of soluble (rather than insoluble) dietary fiber[64]. Acetate, propionate, and butyrate are the three predominant SCFAs, comprising over 95% of the total, with approximate molar ratios in the colonic lumen of 3:1:1. Meanwhile, intestinal protein catabolism contributes to the SCFA pool through generation of branched-chain fatty acids (specifically isobutyrate, 2-methylbutyrate, and isovalerate), which are entirely derived from catabolism of branched-chain amino acids (valine, isoleucine, and leucine)[65]. Concentrations of SCFAs are highest in the proximal colon and decline distally as they are absorbed by the intestinal epithelium. SCFAs enter the portal circulation and are partially metabolized in the liver before reaching the systemic circulation[66]. Of these, butyrate serves as the primary energy substrate for colonic epithelial cells; propionate contributes to the regulation of protein synthesis and hepatic gluconeogenesis; and acetate functions as a substrate for cholesterol and fattyacid synthesis. Reported mean peripheral plasma concentrations are approximately 25-250 μM for acetate, 1.4-13.4 μM for propionate, and 0.5-14.2 μM for butyrate[67].
The biological effects of SCFAs are predominantly mediated through two signaling cascades: activation of G protein-coupled receptors (GPCRs) and inhibition of histone deacetylases (HDACs). The principal SCFA-responsive GPCRs include GPR41 (FFA3), GPR43 (FFA2), GPR109A, and the olfactory receptor 78 (Olfr78). GPR41 is widely expressed in various tissues, while GPR43 is highly expressed in lymphoid tissues and immune cells, both of which respond to acetate, propionate, and butyrate. GPR109A is more sensitive to butyrate[68]. Histone acetylation and deacetylation dynamically govern chromatin structure and gene transcription. Histone acetyltransferases (HATs) catalyze acetyl transfer to lysine residues, promoting open chromatin and transcriptional activation, whereas HDACs remove acetyl groups, leading to chromatin condensation and transcriptional repression[69]. SCFAs, especially butyrate and propionate, are potent HDAC inhibitors that raise histone acetylation levels, which in turn regulate the expression of inflammatory mediators, antioxidant defense systems, and metabolic regulatory genes. It is worth noting that the heterogeneous distribution of SCFAs throughout the body, as well as differences in the expression profiles of SCFA receptors and HDACs across cell types, jointly determine the significant context-dependent effects of SCFAs observed under both homeostatic and pathological conditions[68] [Figure 3].
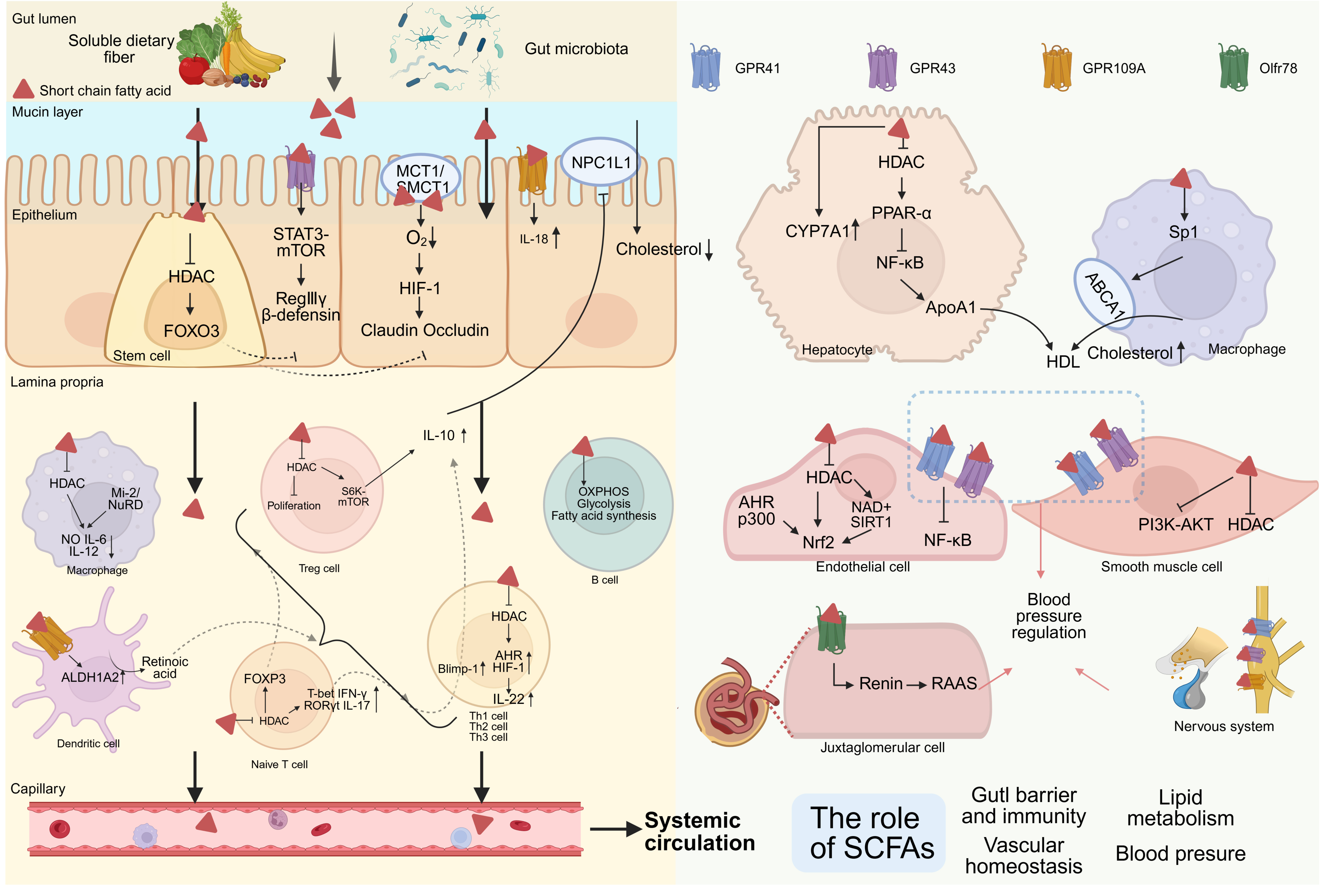
Figure 3. Roles of Short-chain fatty acids (SCFAs). SCFAs counter vascular aging by enhancing intestinal barrier integrity, modulating mucosal immunity, regulating lipid metabolism, and improving endothelial and vascular smooth muscle cell (VSMC) function. They also help maintain blood-pressure homeostasis by coordinating endothelium-smooth muscle vasomotor responses in resistance vessels, modulating renin release and nervous system. (Created in BioRender. Raitasi, E. (2026) https://BioRender.com/8wwkm3n). MCT1: Monocarboxylate transporter 1; SMCT1: sodium-coupled monocarboxylate transporter 1; mTOR: mammalian target of rapamycin; STAT3: signal transducer and activator of transcription 3; HIF-1: hypoxia-inducible factor-1; NPC1L1: niemann-pick-c1-like-1; HDAC: histone deacetylase; FOXO3: forkhead box O3; NuRD: nucleosome remodeling and deacetylase; S6K: S6 kinase; OXPHOS: oxidative phosphorylation; ALDH1A2: aldehyde dehydrogenase 1 family member A2; FOXP3: forkhead box P3; T-bet: T-box expressed in T cells; IFN-γ: interferon-γ; RORγt: retinoic acid receptor-related orphan receptor γt; Blimp-1: B-lymphocyte-induced maturation protein-1; AHR: aryl hydrocarbon receptor; CYP7A1: cholesterol 7α-hydroxylase; PPAR-α: peroxisome proliferator-activated receptor-α; NF-κB: nuclear factor-κB; ApoA1: apolipoprotein A1; Sp1: specificity protein 1; ABCA1: ATP-binding cassette transporter A1; SIRT1: silent information regulator 1; Nrf2: nuclear factor erythroid 2-related factor 2; PI3K: phosphatidylinositol 3-kinase; Akt: protein kinase B; RAAS: renin angiotensin aldosterone system.
At the systemic level, SCFAs exert anti-vascular-aging effects through local protection, regulation of metabolic homeostasis, and improvement of EC and VSMC function. In intestinal epithelial cells, butyrate promotes phosphorylation of mammalian target of rapamycin (mTOR) and signal transducer and activator of transcription 3 (STAT3) via activation of GPR43, driving upregulation of antimicrobial peptide expression (β-defensins, RegIIIγ)[70] and promoting metabolic reprogramming from glycolytic toward fatty acid oxidative. This metabolic transition increases mitochondrial oxygen consumption, establishes physiological hypoxic microenvironments, and stabilizes hypoxia-inducible factor-1 (HIF-1), culminating in enhanced epithelial barrier function[68,71]. In vitro investigations show that SCFAs upregulate tight junction protein expression including claudin-1, occludin, and zonula occludens-1, through STAT3 and specificity protein 1 (Sp1) transcription factor activation, strengthening the junctions. Concurrently, butyrate suppresses "leaky" tight junction protein claudin-2 expression through IL-10 receptor-dependent signaling pathways, reducing non-selective paracellular ion and fluid translocation and further consolidating barrier integrity[72]. In intestinal stem cells, butyrate inhibits HDAC3 and enhances FOXO3 promoter transactivation, which suppresses excessive proliferation, reduces OS and genotoxic damage, and maintains epithelial renewal balance[73]. Acetic acid and propionic acid can also promote Paneth cells to produce Wnt3 signals, promoting the proliferation and differentiation of intestinal stem cells. The increased goblet cells can protect the intestinal tract by secreting mucin[74]. SCFAs modulate the mucosal immune system by coordinating the interaction between innate immunity and adaptive immunity, establishing a protective intestinal immune microenvironment. Butyrate can inhibit HDACs and induce Mi-2 expression, which enhances the binding activity of the Mi-2/nucleosome remodeling and deacetylase (Mi-2/NuRD) complex on the DNA of secondary response genes. This progress downregulates the expression of pro-inflammatory mediators in macrophages induced by lipopolysaccharide (LPS), such as NO, IL-6, and IL-12[68,75]. Additionally, butyrate can promote the secretion of retinoic acid (RA) by colonic macrophages or dendritic cells through GRP109A and enable them to induce differentiation of regulatory T (Treg) cells and IL-10-producing T cells. Moreover, GPR109A was essential for butyrate-mediated induction of IL-18 in colonic epithelium[76]. SCFAs contribute to the proliferation and immunosuppressive capacity of Treg cells, an effect mediated through activation of GPR43 and inhibition of HDAC[77]. Butyrate appears to play a particularly prominent role, facilitating Treg differentiation through enhanced histone acetylation at the forkhead box P3 (FOXP3) promoter as well as at conserved non-coding elements within the FOXP3 locus[78]. In the regulation of T helper (Th) cell subsets, butyrate has differential effects on Th1 and Th17 cells: butyrate promotes Th1 cell differentiation by up-regulating T-box expressed in T cells (T-bet) and interferon-γ (IFN-γ) expression, and inhibits Th17 cell differentiation by suppressing IL-17 and retinoic acid receptor-related orphan receptor γt (RORγt) expression[79]. Meanwhile, butyrate can induce IL-10 production in T cells during Th1 and Th17 differentiation, a process associated with the up-regulation of B-lymphocyte-induced maturation protein-1 (Blimp-1)[79].In Treg cells, SCFAs induce IL-10 via the increased of acetylation of p70 S6 kinase (p70 S6K) and the downstream phosphorylation of the ribosomal protein, which increase mTOR activity as required for IL-10 production[80]. In CD4+ T cells, SCFAs can promote the production of IL-22 by up-regulating the expression of aryl hydrocarbon receptor (AHR) and HIF-1, a process that is differentially regulated by STAT3 and mTOR. SCFAs can enhance the binding ability of HIF-1 to the IL-22 promoter through histone modification[81]. In B cells, SCFAs promote the differentiation of B cells into plasma cells and enhance antibody production by promoting oxidative phosphorylation, glycolysis and fatty acid synthesis. In addition, SCFAs can also participate in class switch recombination by inducing histone acetylation at the immunoglobulin heavy chain locus[82]. These mechanisms collectively attenuate intestinal-derived inflammation and functional integrity of the vascular wall mediated by portal vein-systemic circulation of LPS and other toxic microbial metabolites, indirectly delaying the process of vascular aging.
At the preclinical level, supplementation with acetate, propionate, or butyrate significantly reduces total cholesterol, non-HDL-c, and the non-HDL-c/HDL-c ratio in hamsters[83]. In humans with hypercholesterolemia, oral propionate decreases total cholesterol by approximately 7.3%, LDL-c by 8.1%, and non-HDL-c by 9.1%[84]. SCFAs can lower cholesterol via multiple mechanisms. On one hand, butyrate in particular suppresses hepatic HDAC to activate PPAR-α and upregulate carnitine palmitoyltransferase 1 (CPT1), which concurrently inhibits NF-κB-mediated inflammation and relieves the transcriptional repression of apolipoprotein A1 (ApoA1), enhancing ApoA1 expression in hepatocytes. ApoA1, a major structural protein of HDL and also a crucial receptor for ATP-binding cassette transporter A1 (ABCA1)-mediated cholesterol efflux, participates in reverse cholesterol transport. Even under simulated gut-liver axis conditions, intestinal lumen-derived butyrate that is absorbed and utilized by intestinal epithelium can reach hepatocytes and maintain its ApoA1-promoting activity. However, its overall effect is attenuated because of epithelial metabolism[85,86]. On the other hand, in ApoE knockout mice, hepatic, aortic, and primary peritoneal macrophage studies show that butyrate, via Sp1-dependent transcriptional activation, significantly upregulates ABCA1, promoting cholesterol efflux to ApoA1 and reverse cholesterol transport, alleviating AS in high fat diet-fed mice[87]. SCFAs also induce CYP7A1 in the liver, promoting cholesterol catabolism to BAs and fecal excretion[85,87]. Regarding cholesterol absorption, propionate induces IL-10 in the gut environment, suppressing Niemann-Pick-C1-like-1 (NPC1L1) expression in the small intestine. This effect can reduce cholesterol absorption and lower serum total cholesterol and LDL-c[84]. Moreover, oral administration of propionate broadly attenuates serum levels of various cholesteryl esters and induces a significant remodeling of the BA pool, characterized by elevated concentrations of glycol-chenodeoxycholic acid (GCDCA), glycol-ursodeoxycholic acid (GUDCA), and deoxycholic acid (DCA). The observed negative correlation between DCA and the reduction in total cholesterol, together with prior evidence that DCA inhibits intestinal cholesterol absorption[88] and activates farnesoid X receptor/Takeda G-protein-coupled receptor 5 (FXR/TGR5) signaling[89], implies that propionic acid may partly mediate its cholesterol-lowering effects by enhancing the production and signaling of secondary BAs, particularly DCA[90]. Through these multilevel regulations, SCFAs play a vital protective role in lowering circulating cholesterol, reducing vascular inflammatory burden, and improving vascular function.
In addition to their systemic metabolic effects, SCFAs can directly regulate vascular wall cells to preserve vascular homeostasis. They predominantly act through activation of GPR41/43 and inhibition of HDAC activity, resulting in suppression of NF-κB signaling and decreased expression of pro-inflammatory cytokines, chemokines, and adhesion molecules[91,92]. In experimental models of diabetic ED, sodium butyrate functions as an HDAC inhibitor that increases H3K9 acetylation at the nuclear factor erythroid 2-related factor 2 (Nrf2) promoter. This epigenetic change, together with AHR and the co-activator p300, is linked to higher Nrf2 mRNA and protein abundance, and has been observed even when Nrf2 nuclear translocation is not measurably enhanced. Elevating Nrf2 typically increases the expression of antioxidant response genes such as NQO1 and HO-1, and this correlates with reduced ROS and lipid peroxidation markers and with partial improvement of ED[93,94]. PAA induces mitochondrial dysfunction and impairs the angiogenic capacity of ECs by generating excessive hydrogen peroxide, promoting EC senescence. Acetate exerts anti-aging effects by restoring NAD+ availability and SIRT1 activity, enhancing Nrf2-driven antioxidant defense, and inhibiting NF-κB-mediated SASP[95]. Under inflammatory or injury-related cues, VSMCs can transition toward a synthetic state, reflected by greater proliferative and migratory capacity and increased ECM production. Consistent with this concept, butyrate inhibits VSMC proliferation and migration, with proposed mechanisms involving HDAC inhibition and reduced phosphatidylinositol 3 kinase-protein kinase B (PI3K-Akt) pathway activity[96].
Hypertension is a clinical condition in which blood flow continuously exerts excessive tensile force on the vessel walls[97]. The accompanying ED, overactivation of the RAAS, oxidative stress, and inflammatory responses all further accelerate arterial stiffness. The reduced buffering effect of stiff arteries leads to greater blood flow shear stress and blood pressure fluctuations, thus forming a vicious cycle between hypertension and arterial stiffness[97]. The SCFA receptors widely expressed in endothelial cells, neurons, and immune cells play a key role in regulating blood pressure. Natarajan et al. reported that deletion of GPR41 leads to elevated systolic blood pressure and pulse pressure, accompanied by an increased heart weight/body weight ratio[98]. Although no blood pressure changes were observed in GPR43 knockout mice[99], human evidence indicates that the expression level of GPR43 in immune cells of hypertensive subjects is decreased, and the expression of GPR41 and GPR43 in immune cells is negatively correlated with human arterial stiffness[100,101]. These findings suggest that the defect in GPR43 signal transduction drives the pro-inflammatory phenotype of hypertensive subjects. Activation of the Olfr78 by propionate on afferent arterioles promotes renin secretion and stimulates the RAAS, eliciting a pressor response[102]. SCFAs modulate both sympathetic and vagal neuronal activity and contribute to cardiovascular control. In addition, SCFAs can act directly within the central nervous system to lower blood pressure[103]. This balanced receptor system, in which GPR41/43 mediates depressor effects and Olfr78 mediates pressor effects, constitutes a sensitive sensory network that perceives SCFA flux from the intestinal lumen and maintains blood pressure homeostasis through opposing mechanisms[104]. Furthermore, emerging evidence indicates that immune cell-intrinsic GPR41/43 signaling activated by SCFAs enhances intestinal barrier function and restricts LPS-TLR4-driven inflammation, protecting the blood vessels from angiotensin II-induced hypertension and associated cardiorenal fibrosis[105]. These findings suggest that targeting the SCFA-GPR41/43-immune axis is a potentially powerful therapeutic strategy for regulating blood pressure and preventing fibrosis.
The mentioned cellular and molecular mechanisms ultimately translate into measurable improvements in macroscopic vascular function, particularly ED and arterial stiffness. In addition to the indirect correlations, there are studies showing a direct association between SCFAs and arterial stiffness. The diversity of the gut microbiota is significantly negatively correlated with the degree of arterial stiffness in women, and women have a greater enrichment of protective bacterial genera that can produce SCFA[59,106]. In patients with T2DM, those with the highest tertile level of butyrate had a lower risk of abnormal Ankle-brachial Index[107]. Meanwhile, additional evidence found that arterial stiffness is associated with reduced expression of GPR41/GPR43 in human circulating immune cells[101]. Although further evidence from interventional animal studies and cohort studies is lacking, the multifaceted effects of SCFAs on the pathogenesis of vascular aging suggest that they may represent a key therapeutic target for the prevention and treatment of vascular aging.
Bile acids
BAs are the main components of bile, which not only promote the absorption of lipids and fat-soluble nutrients in the intestine but also act as signaling molecules to regulate the biosynthesis of BAs and lipid metabolism, glucose homeostasis, and immune responses in the body[108]. The BAs circulating in the body are composed of primary BAs and secondary BAs. Cholesterol is enzymatically hydroxylated by CYP7A1 and sterol 27-hydroxylase (CYP27A1) to generate primary BAs such as cholic acid (CA) and chenodeoxycholic acid (CDCA). After conjugation with taurine or glycine in hepatocytes to form bile salts, these BAs are then secreted into bile and stored in the gallbladder[109,110]. After a meal, the gallbladder contracts to release primary BA salts into the duodenum, helping to emulsify chyme to promote lipid absorption and move along the distal digestive tract. Most of the secreted BAs are absorbed into intestinal epithelial cells through active and passive transport and then reused through the enterohepatic circulation, while the rest are excreted in feces[109,110]. In the colon, the gut microbiota mediates the deconjugation, 7α-dehydroxylation, 6α-hydroxylation, and epimerization of primary BAs to convert them into secondary BAs such as deoxycholic acid (DCA), lithocholic acid (LCA), and ursodeoxycholic acid (UDCA), and most of these BAs also enter the enterohepatic circulation and change the composition of the circulating BA pool[109,110]
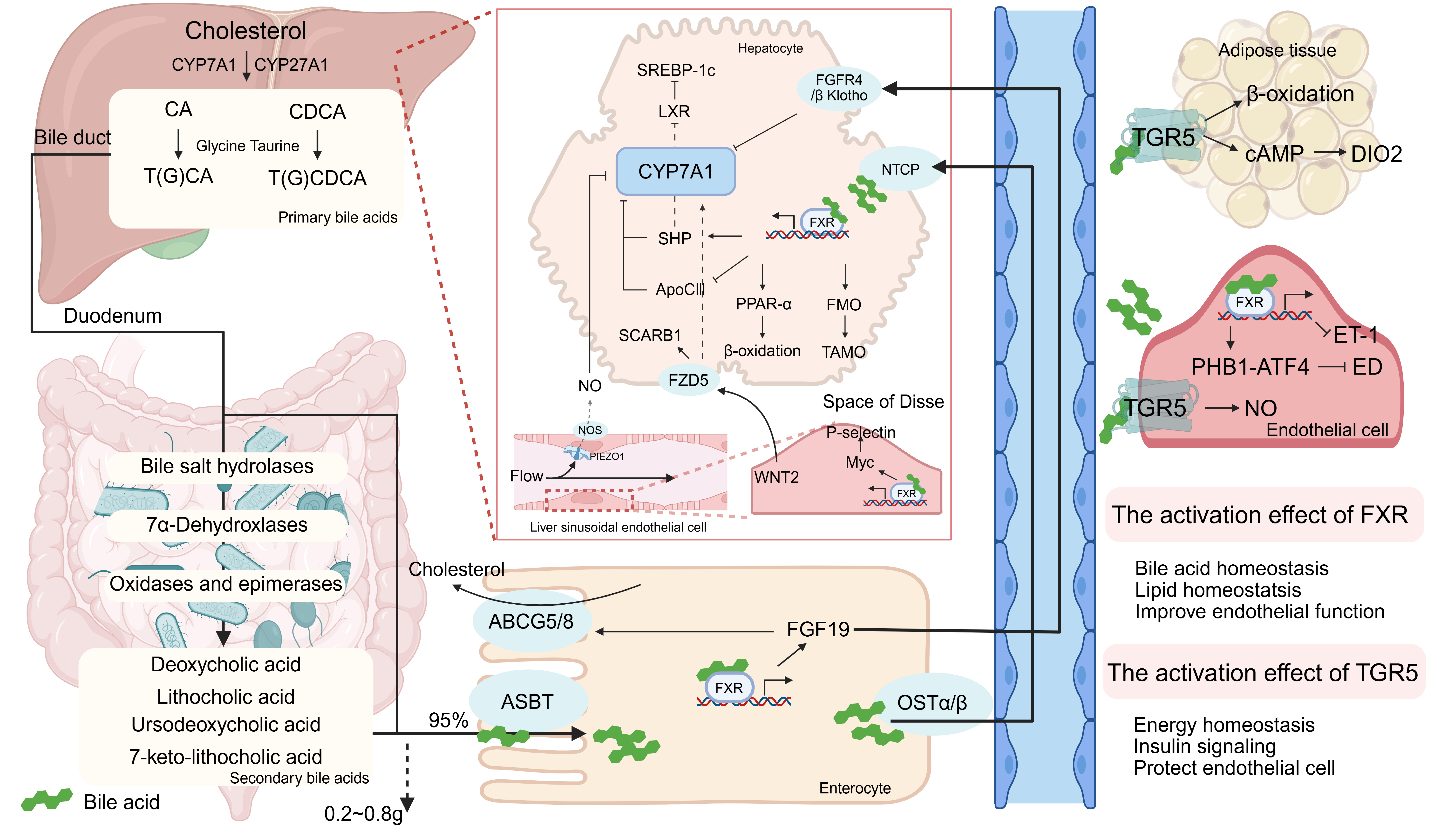
Figure 4. Roles of bile acids (BAs). Primary BAs, synthesized from cholesterol, are converted by gut microbiota into secondary bile acids in the intestinal lumen. Most of primary and secondary BAs traverse the intestinal epithelium and participate in enterohepatic circulation. BAs exert anti-vascular aging effects by modulating lipid and energy metabolism and improving endothelial function. (Created in BioRender. Raitasi, E. (2026) https://BioRender.com/inv9no8). CYP7A1: Cholesterol 7α-hydroxylase; CYP27A1: sterol 27α-hydroxylase; CA: cholic acid; CDCA: chenodeoxycholic acid; ASBT: apical sodium-dependent bile acid transporter;FXR: farnesoid X receptor; TGR5: takeda G-protein-coupled receptor 5; FGF19: fibroblast growth factor 19; ABCG5/8: ATP-binding cassette subfamily G members 5 and 8; OST: organic solute transporter; NTCP: sodium taurocholate cotransporting polypeptide; FGFR4: fibroblast growth factor receptor 4; β Klotho: beta-Klotho; SHP: small heterodimer partner; LXR: liver X receptor; SREBP-1c: sterol regulatory element binding protein-1c; ApoCIII: apolipoprotein CIII; PPAR: peroxisome proliferator-activated receptor; FMO: flavin-containing monooxygenase; TMAO: trimethylamine N-oxide; PIEZO1: PIEZO-type mechanosensitive ion channel component 1; NOS: NO synthase; WNT2: wingless-type MMTV integration site family member 5; FZD5: frizzled class receptor 5; SCARB1: scavenger receptor class B member 1; DIO2: iodothyronine deiodinase 2; ET-1: endothelin-1; PHB-1: prohibitin-1; ATF4: activating transcription factor 4; ED: endothelial dysfunction.
BAs exert their biological effects primarily through multiple receptor families including nuclear receptors such as FXR, pregnane X receptor (PXR), and vitamin D receptor (VDR), as well as the G protein-coupled receptor (GPCR) TGR5[111]. The distribution of BA receptors in tissues is extensive including in the gallbladder, intestines, liver, adipose tissue, brain, and heart. Most of these receptors are expressed in multiple pathological states of cardiovascular tissues, suggesting that BAs may play an important regulatory role in CVDs[111]. Among BA-responsive receptors, FXR and TGR5 have received particular attention because of their established links to metabolic control. FXR is enriched in the liver and intestine, and CDCA is generally considered a strong endogenous FXR agonist. TGR5 is prominently expressed in adipose tissue and other peripheral sites, and several secondary BAs show comparatively higher activity toward TGR5 in commonly used assays. Conjugation of secondary BAs with either taurine or glycine further enhances TGR5 activity. Therefore, BAs exert their corresponding effects as ligands of FXR and TGR5, and the intensity of their actions depends not only on the concentrations of different BAs in the body but also on the density of the above-mentioned receptors in the target tissues[108].
Multiple studies have shown that BAs can affect the function and homeostasis of the intestine. Obesity increases the level of DCA in the ileum, which continuously activates the FXR signaling pathway in intestinal epithelial cells, leading to Paneth cell dysfunction and affecting the function of stem cells[112]. This conclusion supplements the role of activating intestinal FXR in restricting the malignant proliferation of intestinal stem cells[113] and the role of activating TGR5 in promoting the proliferation and reconstruction of stem cells[114]. Long-term use of aspirin can cause gastrointestinal damage. The transformation product of CDCA, 7-keto-lithocholic acid (7-keto-LCA), can effectively repair the intestinal mucosal barrier. It inhibits the intestinal FXR receptor, activates the Wnt signaling pathway, and promotes the self-renewal of intestinal stem cells and epithelial repair[115]. CA can inhibit PPAR-α-mediated fatty acid oxidation (FAO), leading to the obstruction of intestinal stem cell renewal[116]. In addition, activating intestinal FXR can reduce intestinal barrier damage and bacterial translocation in animal models of liver cirrhosis[117]. In terms of inflammation, LCA and DCA and their derivatives can inhibit the differentiation of pro-inflammatory T helper 17 (TH17) cells by promoting Treg cells in an FXR-independent manner, reducing intestinal inflammation. While the stimulation of DCA on macrophages can polarize them to the M1 type, which promotes inflammation[118]. LCA can also block NLRP3 inflammasome-dependent inflammation in bone-marrow-derived macrophages and mice by activating the TGR5-cyclic adenosine monophosphate-protein kinase A (TGR5-cAMP-PKA) pathway[119]. Recent research indicates that ursodeoxycholic acid supplementation enhances secretion of glucagon-like peptide-1 (GLP-1) from enteroendocrine cells through FXR inhibition, mitigating osteoarthritis and extending the anti-inflammatory actions of BAs beyond the gastrointestinal tract[120].
BAs affect chronic vascular inflammation by regulating lipid metabolism. FXR-deficient mice display a characteristic disturbance in BA and lipid processing including higher circulating BAs, cholesterol, and triglycerides, hepatic lipid accumulation, and a more proatherogenic serum lipoprotein profile. These phenotypes support an important role for FXR in maintaining BA-lipid homeostasis[121]. BAs returning to the liver through the portal circulation activate FXR in hepatocytes, which in turn coordinates lipid metabolism via several transcriptional programs. Farnesoid X receptor (FXR) binds and initiates the expression of the SHP gene, generating a large amount of small heterodimer partner (SHP) protein. The latter, as a co-repressor, affects lipid metabolism by reducing the expression of CYP7A1 and the key factor for fat synthesis, sterol regulatory element binding protein-1c (SREBP-1c)[122]. FXR activation has also been associated with lower apolipoprotein CIII (ApoCIII) expression, consistent with improved clearance of triglyceride-rich lipoproteins[123]. In addition, FXR can intersect with PPAR-α pathways that favor fatty-acid β-oxidation and limit hepatic and circulating triglyceride accumulation[124]. At the intestinal level, activation of the fibroblast growth factor 19 (FGF19) pathway promotes the formation of a more hydrophilic bile salt pool and simultaneously upregulates ATP-binding cassette subfamily G members 5 and 8 (ABCG5/8), which enhance cholesterol secretion into the intestinal lumen[125]. Suppression of either BSH-expressing gut microbiota or intestinal FXR-FGF19 signaling relieves the repression of CYP7A1, which augments hepatic BA synthesis and elevates fecal BA excretion, reducing hepatic and systemic cholesterol levels[126]. High BSH-expressing gut microbiota substantially reshapes the host BA profile by reducing circulating tauro-β-muricholic acid (TβMCA), an endogenous FXR antagonist, changing FXR signaling, and ultimately decreasing serum cholesterol and hepatic triglycerides[127].
Tissue-specific FXR knockout studies reveal that FXR-mediated lipid regulation exhibits pronounced tissue-dependent effects. Hepatic FXR activation suppresses lipid synthesis-related gene expression through an SHP- and SREBP-1c-independent pathway, reducing monounsaturated fatty acid levels. Selective activation of intestinal FXR primarily functions by restraining intestinal lipid absorption, especially the uptake of polyunsaturated fatty acids, improving systemic lipid profiles through a distinct anatomical mechanism[128]. Additionally, recent investigations have uncovered a novel mechanism for BA-mediated lipid homeostasis: a reduction in total BAs, specifically conjugated primary BAs, selectively impairs the absorption of saturated fatty acids while comparatively maintaining that of polyunsaturated fatty acids[129]. This shift is accompanied by enhanced secretion of appetite-suppressing hormones and a concurrent restraint on excessive caloric intake, which contributes to the prevention of obesity[129]. The differences in FXR regulation in mediating lipid metabolism and related pathophysiological processes may be attributed to its distinct roles in different tissues as well as other confounding factors including dietary patterns and treatment conditions.
TGR5 is another key BA receptor and can complement FXR in shaping systemic lipid and energy metabolism. DCA administration significantly reduces postprandial lipemia including cholesterol and triglycerides. Critically, this lipid-lowering effect persists in FXR-knockout mice, indicating that DCA acts predominantly through TGR5 rather than FXR[89]. In white adipose tissue, TGR5 signaling has been linked to mitochondrial fission and “beige” remodeling, together with increased lipolysis and availability of free fatty acids, changes that are compatible with enhanced fatty-acid β-oxidation and heat production[130]. In rodent models, BAs activate the TGR5-cAMP-iodothyronine deiodinase 2 (DIO2) signaling pathway in brown adipose tissue, which increases thyroid hormone activation and is associated with improved energy expenditure and metabolic phenotypes[131]. Clinical evidence further demonstrates that administration of CDCA promotes brown adipocyte expansion in humans and raises energy expenditure through a TGR5-cAMP-DIO2-dependent mechanism[132]. Long-term statin administration is associated with an increased risk of impaired glucose tolerance. Clinical studies indicate that cosupplementation with UDCA alleviates above effect without compromising the lipidlowering efficacy of statins. The principal mechanism involves UDCA functioning as a TGR5 ligand, which stimulates GLP-1 secretion from enteroendocrine cells and contributes to the maintenance of glucose homeostasis[133].
Beyond their systemic metabolic effects, BAs also exert direct effects on vascular cells via FXR and TGR5 signaling. In rat pulmonary arterial ECs, FXR activation suppresses endothelin-1 (ET-1) promoter activity as well as heterologous promoters driven by activator protein 1 (AP-1) response elements[134]. Subsequent studies show that in cholestasis and chronic liver disease, elevated circulating BAs suppress ET-1 production, which may underlie systemic vasodilation[135]. TGR5 activation also induces endothelial NO generation through Akt phosphorylation and increased intracellular Ca2+, inhibiting monocyte adhesion and attenuating vascular inflammation[136]. BAs also influence vascular health through the trimethylamine N-oxide (TMAO) pathway. FXR regulates flavin-containing monooxygenase 3 (FMO3) expression and activity, promoting trimethylamine (TMA) conversion to TMAO, a mediator of vascular toxicity[137,138]. Recent studies suggest CDCA as a biomarker for obesity-related ED, whereas tauro-chenodeoxycholic acid (TCDCA) demonstrates therapeutic potential in alleviating obesity-related ED through the FXR-prohibitin1-activating transcription factor 4 (FXR-PHB1-ATF4) pathway, which upregulates serine and one-carbon metabolism[139]. These findings suggest novel therapeutic strategies for preventing vascular aging-related diseases. At present, the direct influence of BAs on arterial stiffness mainly remains at the level of cross-sectional studies, for example, fecal microbiota transplantation from patients with coronary artery disease into germ-free mice has been shown to induce BA dysregulation and aggravate arterial stiffness[140]. In T2DM model mice, administration of tauro-ursodeoxycholic (TUDCA) acid alleviates arterial stiffness and ED[141]. Based on the establishment of a mouse model of arterial stiffness, intervention studies should be conducted to explore the causal relationship between BAs and arterial stiffness.
Although the effects of BAs on vascular endothelium have gradually been recognized, hepatic sinusoidal endothelium plays a more central role in BA-mediated metabolic regulation[142-144]. Liver sinusoidal endothelial cells (LSECs) are one of the important non-parenchymal cells in the liver. They activate the FZD5 receptor on hepatocyte through paracrine signal WNT2 that regulate various metabolic pathways in the liver[142]. Activation of this axis upregulates genes involved in cholesterol synthesis, transport, and uptake (SCARB1), as well as its conversion into primary BAs (CYP7A1, CYP27A1). More importantly, this pathway enhances the expression of glycine production and bile acid-CoA: amino acid N-acyltransferase (BAAT), resulting in higher levels of glycine-conjugated BAs and an increase in the ratio of glycine/taurine binding in the liver and plasma, which are unique BA composition in humans[142]. Lichtenstein et al. identified a mechanosensitive endothelial pathway that links hepatic blood flow to lipid metabolism[143]. The ion channel PIEZO1 located in LSEC senses blood flow shear force, activates nitric oxide synthase and synthesizes nitric oxide. The latter inhibits the expression of CYP7A1. Consistent with this effect, increased BA levels in the gallbladder and plasma are observed, accompanied by reduced cholesterol levels in both the liver and plasma[143]. Zhang et al. demonstrated that, during cholestasis, CDCA and its conjugate TCDCA activate FXR signaling in LSECs and then induce Myc expression[144]. This upregulation markedly increases P-selectin levels which promotes neutrophil infiltration and exacerbates cholestatic liver injury. These findings uncover a previously unappreciated role for ECs in integrating environmental cues, inflammation, and tissue destruction[144]. LSEC is exposed to higher concentrations of BAs, expresses more related receptors, and forms a tight paracrine regulatory unit with hepatocytes in the perisinusoidal space, directly influencing BA synthesis, transport, and cholesterol metabolism. In comparison, the evidence for the impact of BAs on vascular endothelium is still relatively limited at present. Moreover, a study found that LCA monotherapy mimics the anti-aging benefits of caloric restriction in diverse model organisms and extends lifespan through the activation of AMPK. Nevertheless, its cardiovascular effects require further elucidation[145]. In contrast to SCFAs, BAs exert a more profound regulatory influence on the gut-vascular axis through modulation of intestinal stem cell behavior and metabolic processes. Further investigation is warranted to elucidate the systemic metabolism, tissue distribution of BAs, and the spatial expression patterns of their receptors, which will be critical for determining their direct effects on the vasculature.
Phenylalanine-derived metabolites
Gut microbiota having the porA gene metabolize protein-derived phenylalanine to phenylpyruvate, which is further converted to PAA through two distinct microbial pathways[146]. In the liver, PAA combines with glutamine to form PAGln, which is then excreted through the kidneys. PAGln has multiple clinical uses: serum PAGln serves as an early biomarker of renal impairment and a metabolic indicator in severe hepatic encephalopathy, while urinary PAGln assesses phenylalanine metabolism in phenylketonuria[147].
In a prospective cohort study of patients with advanced chronic kidney disease (CKD), elevated serum PAGln levels were an independent risk factor for all-cause mortality and major cardiovascular events (MACE), suggesting the potential prognostic value of PAGln in predicting cardiovascular outcomes[148]. Nemet and colleagues performed untargeted metabolomic screening in subjects with T2DM and identified PAGln as being strongly associated with major adverse cardiovascular events. This association was validated in an independent cohort of ~4,000 individuals with stable CVDs, and a series of functional experiments supported a causal relationship between PAGln and cardiovascular risk. Importantly, this study first showed that a gut microbiota-derived metabolite promotes platelet hyperresponsiveness and thrombotic potential through adrenergic receptor (ADR) signaling[149]. In male mice chronically exposed to alcohol, gut dysbiosis accompanied by elevated PAGln levels was associated with cardiovascular dysfunction. Further in vitro studies demonstrated that PAGln promotes EC activation, characterized by increased reactive oxygen species and upregulation of pro-inflammatory factors and adhesion molecules. It also inhibits EC migration and wound healing capacity, ultimately contributing to ED[150]. Recent studies have directly linked PAA/PAGln to aging: it acts as a "catecholamine-like" signal and activates the ADR-AMPK pathway, which triggers mitochondrial fragmentation, OS, and DNA damage. Those changes induce senescence in multiple cell types including ECs. In cross-sectional studies of kidney transplant recipients and chronic hemodialysis patients, elevated serum PAGln levels were positively associated with increased c-f PWV[151,152]. Thus, PAA/PAGln functions not only as a cardiovascular risk metabolite but also as a systemic pro-aging signal in various organs[14].
Beyond the metabolites discussed above, the gut microbiota also produces several other molecules that are relevant to vascular function, including LPS, TMAO[153], and indole derivatives[154]. These metabolites influence vascular homeostasis and disease progression through interconnected pathways that involve the gut barrier [155,156], systemic metabolism[157,158], and the vascular microenvironment[159-162].
Arterial stiffness is often accompanied by higher intestinal permeability and elevated circulating LPS levels[60,163]. Under a high-fat diet, TLR4-deficient mice still develop intestinal injury and gut microbiota alterations, but their arterial stiffness is partly alleviated[164]. This finding suggests that LPS-TLR4 signaling plays a key role in this process. Lipopolysaccharide-binding protein (LBP) is an acute-phase reactant. It mediates the interaction between LPS and TLR4 and increases host sensitivity to even very small amounts of bacteria. A study found that the level of LBP was significantly correlated with arterial stiffness in men with T2DM[165]. Dietary choline and carnitine are first converted by gut microbes into TMA, and then oxidized in the liver by FMO3 to form TMAO. In both mice and humans, plasma TMAO levels rise with age[160]. In older adults, circulating TMAO is significantly associated with ED and arterial stiffness[160]. Interventional studies further indicate that exogenous TMAO can induce vascular aging-like phenotypes in young mice, which makes it one of the few microbiota-derived metabolites with causal evidence[166]. Mechanistically, TMAO increases the intrinsic mechanical stiffness of the aorta through the promotion of the formation of advanced glycation end-products (AGEs). Oxidative stress also contributes, at least in part, to TMAO-related arterial stiffening, independent of AGEs[166]. Cross-sectional studies have likewise found that, in patients with end-stage renal disease receiving hemodialysis or peritoneal dialysis, both TMAO levels and age are associated with arterial stiffness[167,168]. In advanced non-dialysis chronic kidney disease, TMAO is associated with peripheral arterial stiffness as well[169]. Tryptophan is an essential amino acid. It not only participates in protein synthesis, but is the precursor for a variety of important bioactive compounds. Gut microbes can metabolize tryptophan into indole and its derivatives, which regulate inflammation, metabolism, and neural function[154]. Although direct evidence linking indole metabolites to arterial stiffness is still lacking, studies have shown that they improve ED in several vascular beds, including the gut, the aorta, and pulmonary[170-172]. This suggests a potential protective role in slowing vascular aging.
Like SCFAs and BAs, these gut-derived metabolites appear to influence vascular aging mainly through indirect regulatory mechanisms. Direct causal evidence remains limited, and their effects differ across populations and disease contexts. Future studies that combine multi-omics analyses with mechanistic experiments are needed to clarify their causal roles in vascular aging.
GUT MICROBIAL METABOLITES IN VASCULAR AGING-RELATED DISEASES
Based on the current understanding of the cellular and molecular mechanisms underlying vascular aging, as well as the regulatory roles of gut microbial metabolites, the following discussion will focus on representative aging-related vascular diseases, particularly AS and aortic aneurysm, to further elucidate the specific contributions of these microbial metabolites to the pathogenesis of these structural vascular lesions
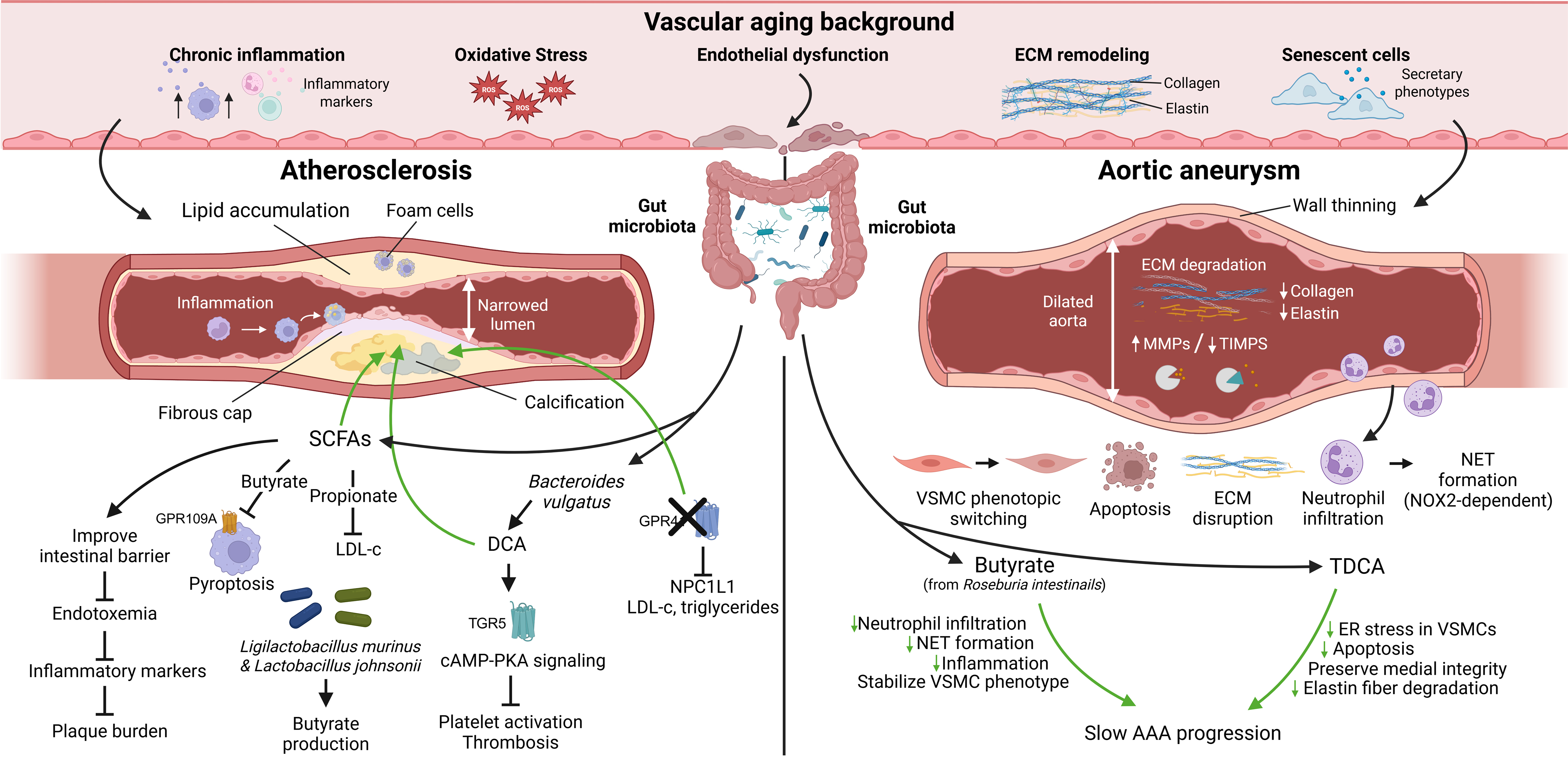
Figure 5. Gut microbial metabolites in vascular aging-related diseases. In the context of vascular aging, gut microbial metabolites can regulate the progression of atherosclerosis and aortic aneurysm through multiple mechanisms. (Created in BioRender. Raitasi, E. (2026) https://BioRender.com/0inu8ug). ECM: Extracellular matrix; SCFAs: short-chain fatty acids; LDL-c: LDL-cholesterol; DCA: deoxycholic acid; cAMP-PKA: cyclic adenosine monophosphate-protein kinase A; NPC1L1: Niemann-Pick-C1-like-1; HDAC: histone deacetylase; MMPs: matrix metalloproteinases; TIMPs: tissue inhibitors of metalloproteinases; NET: neutrophil extracellular trap; TDCA: taurodeoxycholic acid; ER: endoplasmic reticulum.
Atherosclerosis
AS is a chronic arterial disease characterized by the accumulation of lipids, minerals, and cellular debris beneath the intima, forming plaques that cause arterial wall thickening, stiffness, and loss of elasticity[173]. Vascular aging creates a susceptible background for the development of AS through diffuse alterations such as chronic low-grade inflammation, OS, ED, and ECM remodeling[174]. AS manifests as a focal plaque that forms on this aged vascular foundation under the influence of dyslipidemia and various risk factors. In these lesions, continuous production of inflammatory, lipid, and fibrotic mediators occurs, coupled with significant accumulation of senescent cells[24]. Gut microbial metabolites can modulate atherosclerotic susceptibility by regulating these underlying pathological processes, and exert either protective or detrimental effects during plaque formation and progression[52].
Accumulating evidence demonstrates that SCFAs protect against AS. Patients with atherosclerotic cardiovascular disease show reduced butyrate- and propionate-producing bacteria, suggesting cardioprotective roles for SCFAs[175]. For instance, the colonization of butyrate-producing bacterium Roseburia intestinalis ameliorates intestinal barrier function and reduces endotoxemia, decreasing inflammatory markers and atherosclerotic lesion burden in a diet-dependent manner[71]. A clinical cohort study demonstrated that oral propionate supplementation lowers LDL cholesterol levels in hypercholesterolemic subjects, suggesting potential anti-atherosclerotic efficacy[84]. Moreover, Ligilactobacillus murinus and Lactobacillus johnsonii promote butyrate generation through mutualistic interactions with butyrate-producing bacteria. Butyrate then suppresses macrophage pyroptosis via GPR109A, which exerts anti-atherosclerotic effects[176]. BAs also regulate AS development and progression. A recent investigation revealed that patients with coronary atherosclerotic heart disease (CAD) have decreased Bacteroides vulgatus, leading to reduced circulating DCA levels that weaken platelet TGR5-cAMP-PKA signaling and enhance Ca2+-protein kinase C (PKC) pathway activity, ultimately resulting in platelet hyperresponsiveness, increased thrombosis, and myocardial injury. Supplementation with DCA, B. vulgatus, or fecal microbiota transplantation (FMT) from healthy donors reversed these effects[177]. Fibrinolytic capacity is weakened in obese mice, a phenomenon primarily attributable to obesity-induced metabolic stress that attenuates hepatic FXR signaling, alleviating its transcriptional repression of the PAI-1 gene and exacerbating thrombotic propensity. Administration of FXR agonists reverses this pathological condition in obese mice[178]. The SCFA receptor GPR41, although not directly determining atherosclerotic plaque burden, functions as a critical intestinal chemoreceptor in male mice. Its deletion substantially remodels the gut microbiota-BA axis, reducing secondary BAs and FXR target gene expression, and downregulating the ileal cholesterol transporter NPC1L1 and nutrient transporters. This leads to decreased LDL cholesterol, triglycerides, adipose mass, and liver weight, manifesting a systemic anti-hypercholesterolemic phenotype with potential anti-atherosclerotic activity[179].
Aortic aneurysm
Epidemiological evidence demonstrates that increased arterial age is independently associated with accelerated thoracic aortic aneurysm (TAA) growth, linking aortic aneurysm natural history to vascular aging[180]. Aortic aneurysm is a permanent focal dilation of the aorta, with a diameter ≥ 50% larger than normal. Despite existing pathophysiological differences, TAA and AAA share several features: compared with normal aorta, aneurysmal wall demonstrates decreased elastin, collagen, glycosaminoglycans, and an imbalance between MMPs and tissue inhibitors of metalloproteinases (TIMPs)[181]. Notably, similar matrix degradation and remodeling processes occur during the natural aging of the aorta itself, leading investigators to propose that TAA may represent a focal, accelerated form of aortic aging in some patients. Thus, targeting VSMC phenotypic switching, apoptosis, and ECM disruption to slow aneurysm progression has become a research priority[46,182].
Identifying gut microbial metabolites that suppress aberrant VSMC phenotypic switching is clinically significant. In AAA mouse models, Roseburia intestinalis and its metabolite butyrate substantially reduce neutrophil infiltration and NOX2-dependent neutrophil extracellular trap (NET) formation, attenuating inflammation and aberrant VSMC phenotypic switching, which slows AAA progression[183]. Another study reported that taurodeoxycholic acid (TDCA) reduced endoplasmic reticulum stress in VSMCs and decreased apoptosis. In parallel, TDCA preserved medial integrity and limited elastic fiber degradation, suppressing the formation and progression of AAA[184]. These observations support the concept that SCFAs and BAs may influence aneurysm susceptibility, particularly AAA, by regulating VSMC phenotype and viability. Further studies are needed to identify other metabolites, more thoroughly clarify the underlying mechanisms, and compare the similarities and differences in mechanistic pathways between TAA and AAA.
RESEARCH GAP AND PROSPECTS
By incorporating and summarizing the relevant literature on the gut microbiota and vascular aging, this review found that the evidence provided by the original studies still has several limitations. First, studies exploring the molecular basis of vascular aging at the endothelial level often rely on “readout”-type descriptors such as reduced NO bioavailability, increased ROS, and upregulation of adhesion molecules, without firmly establishing the temporal sequence and causal links to clinically quantifiable vascular aging phenotypes like arterial stiffening, pulse wave velocity, and altered vascular compliance. In addition, the heterogeneity of endothelial responses across vascular beds and distinct biomechanical milieus (shear stress and pulsatile stretch) is frequently underappreciated. Second, VSMC phenotypic switching is widely viewed as a key driver of vascular remodeling, ECM reorganization, calcification, and stiffening. However, nomenclature and marker systems used to define VSMC states have not yet been fully standardized, and quantitatively comparable estimates of the stage-specific contribution of VSMCs to stiffening, ECM remodeling, and calcification are still lacking. Third, research on immune cell infiltration remains largely phenomenological, focusing on changes in immune cell abundance and inflammatory mediators, with limited mechanistic insight into which immune subsets, at which stages, engage specific ligand-receptor interactions to program endothelial and VSMC aging. Fourth, current work disproportionately centers on a small set of “signature” metabolites, whereas broader microbiota-derived metabolomic networks and metabolite-metabolite interactions have not been systematically mapped, hampering an integrated understanding of the complex relationships between metabolite landscapes and vascular aging phenotypes. Finally, the evidence linking microbial metabolites to vascular aging is still dominated by animal studies and cross-sectional human investigations. Analytical studies and interventional trials with stronger causal inference capabilities are still scarce, especially those with vascular aging phenotypes as the primary clinical endpoints.
Looking ahead, mechanistic studies should be based on animal and cellular models that more realistically simulate the process of human vascular aging, and integrate multi-omics approaches including metagenomics, metabolomics, single-cell transcriptomics, and spatial transcriptomics. Focusing on the proposed “microbiota-metabolite-receptor-signaling pathway-vascular phenotype” axis, future work should delineate how specific microbial metabolites modulate aging phenotypes of ECs, VSMCs, and immune cells, as well as their local microenvironment. At the same time, heterogeneity in different vascular beds and at different disease stages, as well as causal validation of key points, should also be emphasized. In terms of clinical translation, the field requires prospective cohort studies and mechanism-guided interventional trials. On the basis of unifying the phenotypes of vascular aging, such studies must establish the above-mentioned phenotypes as the primary endpoints to comprehensively evaluate the clinical potential of specific microbial metabolites and their signaling cascades. In conclusion, incorporating gut microbial metabolites into the research on vascular aging will help to more comprehensively reveal the pathogenesis of CVDs and provide new strategies for prevention and intervention by reshaping the local metabolic microenvironment.
CONCLUSIONS
Vascular aging represents a complex process driven by the interaction of ED, VSMC phenotypic switching, oxidative stress and so on. Substantial evidence emphasizes the critical role of gut microbial-derived metabolites such as SCFAs, BAs, and phenylalanine-derived metabolites in modulating these processes through diverse molecular pathways. These metabolites link the intestinal microbiota and vascular homeostasis and diseases by the influence of signaling networks related to immune regulation, metabolic balance, oxidative stress and inflammation. The important point is that they are both protective and pathogenic factors which highlight the complexity and context-dependency of the interaction between the host and the microbial community. Although the current researches have provided compelling insights into the gut-vascular axis, there are still significant gaps in establishing causal relationships, clarifying mechanism specificity, and translating experimental results into clinical applications. Future research will integrate multi-omics methods, intervention studies and functional knockout experiments to fully address these issues. Ultimately, a deeper understanding of the metabolic products of gut microbiome may provide a new perspective for precisely preventing and treating vascular aging and its associated diseases.
DECLEARATIONS
Acknowledgments
The graphical abstract was created with BioRender.com (Created in BioRender. Raitasi, E. (2026) https://BioRender.com/18hm9fk).
Authors’ contributions
Determined the theme and research framework: Li T (Tianci Li), Li T (Ting Li)
Searched for, screened and organized literature: Hua Y, Cao Y, Sun H
Wrote the manuscript: Li T (Tianci Li)
Created 3, 1 and 1 BioRender figures, respectively: Li T (Tianci Li), Hua Y, Cao Y
Revised the manuscript: Li T (Ting Li), Yuan Z
All the authors edited the manuscript and approved the final manuscript.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool GPT (version 5.4 mini, constant iteration) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by the following funding sources: National Key R&D Program of China (2021YFA1301201, 2021YFA0805400, 2024YFA1307004); National Natural Science Foundation of China (Nos. 82370458, 82430019, 82570531).
Conflict of interest
Li T (Ting Li) is an Editorial Board Member of The Journal of Cardiovascular Aging and also serves as a Guest Editor for the Special Issue “Gut Microbiota and Metabolites in Cardiovascular Aging and Disease”. Li T (Ting Li) had no involvement in the editorial processing of this manuscript, including reviewer selection, manuscript handling, or decision-making, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. United Nations. World population prospects 2024: summary of results. Available from: https://desapublications.un.org/publications/world-population-prospects-2024-summary-results [Last accessed on 26 May 2026].
2. Ghosh TS, Shanahan F, O’toole PW. The gut microbiome as a modulator of healthy ageing. Nat Rev Gastroenterol Hepatol. 2022;19:565-84.
3. He S, Huang B, Xu F, et al. Functions and application of circRNAs in vascular aging and aging-related vascular diseases. J Nanobiotechnol. 2025;23:216.
4. Witkowski M, Weeks TL, Hazen SL. Gut microbiota and cardiovascular disease. Circ Res. 2020;127:553-70.
5. Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. 2020;19:55-71.
6. Lynch SV, Pedersen O. The human intestinal microbiome in health and disease. N Engl J Med. 2016;375:2369-79.
8. Ling Z, Liu X, Cheng Y, Yan X, Wu S. Gut microbiota and aging. Crit Rev Food Sci Nutr. 2020;62:3509-34.
9. Wilmanski T, Diener C, Rappaport N, et al. Gut microbiome pattern reflects healthy ageing and predicts survival in humans. Nat Metab. 2021;3:274-86.
10. Francini E, Badillo Pazmay GV, Fumarola S, Procopio AD, Olivieri F, Marchegiani F. Bi-directional relationship between bile acids (BAs) and gut microbiota (GM): UDCA/TUDCA, probiotics, and dietary interventions in elderly people. Int J Mol Sci. 2025;26:1759.
11. Biagi E, Franceschi C, Rampelli S, et al. Gut microbiota and extreme longevity. Curr Biol. 2016;26:1480-5.
12. Longtine AG, Greenberg NT, Gonzalez A, et al. Oral supplementation with the short-chain fatty acid acetate ameliorates age-related arterial dysfunction in mice. Aging Biol. 2024;2:20240033.
13. Malik M, Suboc TM, Tyagi S, et al. Lactobacillus plantarum 299v supplementation improves vascular endothelial function and reduces inflammatory biomarkers in men with stable coronary artery disease. Circ Res. 2018;123:1091-102.
14. Yang H, Wang T, Qian C, et al. Gut microbial-derived phenylacetylglutamine accelerates host cellular senescence. Nat Aging. 2025;5:401-18.
15. Chen W, Li M, Zeng G, et al. Gut microbiota-derived metabolite phenylacetylglutamine in cardiovascular and metabolic diseases. Pharmacol Res. 2025;217:107794.
16. Chen B, Wirawan KF, Luo L, Zhang J, Li T. Mapping arterial stiffness metabolic biomarkers: a bibliometric analysis. Front Med. 2025;12:1557731.
17. Ungvari Z, Tarantini S, Sorond F, Merkely B, Csiszar A. Mechanisms of vascular aging, a geroscience perspective: JACC focus seminar. J Am Coll Cardiol. 2020;75:931-41.
18. Couteur DG, Lakatta EG. A vascular theory of aging. J Gerontol A Biol Sci Med Sci. 2010;65A:1025-7.
19. Grunewald M, Kumar S, Sharife H, et al. Counteracting age-related VEGF signaling insufficiency promotes healthy aging and extends life span. Science. 2021;373:eabc8479.
20. Lacolley P, Avril S, Gáll T, et al. Aging in the vascular system: lessons from mechanobiology, computational approaches, and oxidative stress. Cardiovasc Res. 2025;121:1566-81.
21. Climie RE, Alastruey J, Mayer CC, et al. Vascular ageing: moving from bench towards bedside. Eur J Prev Cardiol. 2023;30:1101-17.
22. Wang M, Monticone RE, Mcgraw KR. Proinflammatory arterial stiffness syndrome: a signature of large arterial aging. J Vasc Res. 2018;55:210-23.
23. Donato AJ, Machin DR, Lesniewski LA. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ Res. 2018;123:825-48.
24. Herzog MJ, Müller P, Lechner K, et al. Arterial stiffness and vascular aging: mechanisms, prevention, and therapy. Sig Transduct Target Ther. 2025;10:282.
25. Hooglugt A, Klatt O, Huveneers S. Vascular stiffening and endothelial dysfunction in atherosclerosis. Curr Opin Lipidol. 2022;33:353-63.
26. Regnault V, Lacolley P, Laurent S. Arterial stiffness: from basic primers to integrative physiology. Annu Rev Physiol. 2024;86:99-121.
27. Li Q, Qian Z, Huang Y, et al. Mechanisms of endothelial senescence and vascular aging. Biogerontology. 2025;26:128.
28. Xu S, Ilyas I, Little PJ, et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev. 2021;73:924-67.
29. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263-71.
30. Carnagarin R, Matthews V, Zaldivia MTK, Peter K, Schlaich MP. The bidirectional interaction between the sympathetic nervous system and immune mechanisms in the pathogenesis of hypertension. Br J Pharmacol. 2018;176:1839-52.
31. Drüppel V, Kusche‐vihrog K, Grossmann C, et al. Long‐term application of the aldosterone antagonist spironolactone prevents stiff endothelial cell syndrome. FASEB J. 2013;27:3652-9.
32. Peng X, Haldar S, Deshpande S, Irani K, Kass DA. Wall stiffness suppresses Akt/eNOS and cytoprotection in pulse-perfused endothelium. Hypertension. 2003;41:378-81.
33. Méndez-Barbero N, Gutiérrez-Muñoz C, Blanco-Colio L. Cellular crosstalk between endothelial and smooth muscle cells in vascular wall remodeling. Int J Mol Sci. 2021;22:7284.
34. Liu X, Liu H, Lin Y, et al. Deletion of METTL14, a key methylation regulator, attenuates vascular ageing. Eur Heart J. 2025;46:4953-68.
35. Pang J, Pan L, Shen W, et al. Endothelial LRRC8A delays vascular ageing in natural and accelerated ageing mouse models. Cardiovasc Res. 2025;121:2549-64.
36. Cao G, Xuan X, Hu J, Zhang R, Jin H, Dong H. How vascular smooth muscle cell phenotype switching contributes to vascular disease. Cell Commun Signal. 2022;20:180.
37. Qiu H, Zhu Y, Sun Z, et al. Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res. 2010;107:615-9.
38. Badi I, Mancinelli L, Polizzotto A, et al. miR-34a promotes vascular smooth muscle cell calcification by downregulating SIRT1 (Sirtuin 1) and Axl (AXL receptor tyrosine kinase). Arterioscler Thromb Vasc Biol. 2018;38:2079-90.
39. Chen L, Fukuda N, Otsuki T, et al. Increased complement 3 with suppression of miR‐145 induces the synthetic phenotype in vascular smooth muscle cells from spontaneously hypertensive rats. J Am Heart Assoc. 2019;8:e012327.
40. Yuan X, Bhat OM, Samidurai A, Das A, Zhang Y, Li P. Reversal of endothelial extracellular vesicle-induced smooth muscle phenotype transition by hypercholesterolemia stimulation: role of NLRP3 inflammasome activation. Front Cell Dev Biol. 2020;8:597423.
41. Yan W, Li T, Yin T, et al. M2 macrophage-derived exosomes promote the c-KIT phenotype of vascular smooth muscle cells during vascular tissue repair after intravascular stent implantation. Theranostics. 2020;10:10712-28.
42. Grootaert MO, Finigan A, Figg NL, Uryga AK, Bennett MR. SIRT6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ Res. 2021;128:474-91.
43. Chi C, Fu H, Li Y, et al. Exerkine fibronectin type-III domain-containing protein 5/irisin-enriched extracellular vesicles delay vascular ageing by increasing SIRT6 stability. Eur Heart J. 2022;43:4579-95.
44. Nie H, Ji T, Wan Z, et al. ATF3 deficiency exacerbates ageing‐induced atherosclerosis and clinical intervention strategy. Adv Sci. 2025;12:e02249.
45. Li X, Chen M, Chen X, et al. TRAP1 drives smooth muscle cell senescence and promotes atherosclerosis via HDAC3-primed histone H4 lysine 12 lactylation. Eur Heart J. 2024;45:4219-35.
46. Sun S, Zhang Z, Zhang G, et al. GSDME-dependent pyroptosis drives abdominal aortic aneurysm via promoting vascular senescence. Nat Commun. 2025;16:11248.
47. Sun D, Wu W, Wu J, et al. Pro-ferroptotic signaling promotes arterial aging via vascular smooth muscle cell senescence. Nat Commun. 2024;15:1429.
48. Ajoolabady A, Pratico D, Tang D, Zhou S, Franceschi C, Ren J. Immunosenescence and inflammaging: mechanisms and role in diseases. Ageing Res Rev. 2024;101:102540.
49. Dong R, Ji Z, Wang M, Ma G. Role of macrophages in vascular calcification: from the perspective of homeostasis. Int Immunopharmacol. 2025;144:113635.
50. Deconne TM, Buckley DJ, Trott DW, Martens CR. The role of T cells in vascular aging, hypertension, and atherosclerosis. Am J Physiol Heart Circ Physiol. 2024;327:H1345-60.
51. Pattarabanjird T, Li C, Mcnamara C. B cells in atherosclerosis: Mechanisms and Potential Clinical Applications. JACC Basic Transl Sci. 2021;6:546-63.
52. Fusco W, Adolph T, Cammarota G, Gasbarrini A, Ianiro G, Tilg H. Gut microbiota and atherosclerosis. Gut. 2026;75:1067-77.
53. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14:e1002533.
54. Hul M, Cani PD, Petitfils C, De Vos WM, Tilg H, El-Omar EM. What defines a healthy gut microbiome? Gut. 2024;73:1893-908.
55. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220-30.
56. Odamaki T, Kato K, Sugahara H, et al. Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol. 2016;16:90.
57. Ticinesi A, Maggi S, Nouvenne A, Zuliani G, Franceschi C. The gut microbiome and ageing trajectories: mechanisms and clinical implications. Nat Rev Endocrinol. 2026;22:323-37.
58. Untersmayr E, Brandt A, Koidl L, Bergheim I. The intestinal barrier dysfunction as driving factor of inflammaging. Nutrients. 2022;14:949.
59. Menni C, Lin C, Cecelja M, et al. Gut microbial diversity is associated with lower arterial stiffness in women. Eur Heart J. 2018;39:2390-7.
60. Battson ML, Lee DM, Jarrell DK, et al. Suppression of gut dysbiosis reverses Western diet-induced vascular dysfunction. Am J Physiol Endocrinol Metab. 2018;314:E468-77.
61. Trikha SRJ, Lee DM, Ecton KE, et al. Transplantation of an obesity-associated human gut microbiota to mice induces vascular dysfunction and glucose intolerance. Gut Microbes. 2021;13:1940791.
62. Cuadrat RRC, Goris T, Birukov A, et al. Association of the human gut microbiota with vascular stiffness. Sci Rep. 2023;13:13348.
63. Luo S, Zhao Y, Zhu S, et al. Flavonifractor plautii protects against elevated arterial stiffness. Circ Res. 2023;132:167-81.
64. Rahman S, Trone K, Kelly C, Stroud A, Martindale R. All fiber is not fiber. Curr Gastroenterol Rep. 2022;25:1-12.
65. Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. 2016;165:1332-45.
66. Hu T, Wu Q, Yao Q, Jiang K, Yu J, Tang Q. Short-chain fatty acid metabolism and multiple effects on cardiovascular diseases. Ageing Res Rev. 2022;81:101706.
67. Canfora EE, Jocken JW, Blaak EE. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat Rev Endocrinol. 2015;11:577-91.
68. Mann ER, Lam YK, Uhlig HH. Short-chain fatty acids: linking diet, the microbiome and immunity. Nat Rev Immunol. 2024;24:577-95.
69. Rekha K, Venkidasamy B, Samynathan R, et al. Short-chain fatty acid: an updated review on signaling, metabolism, and therapeutic effects. Crit Rev Food Sci Nutr. 2022;64:2461-89.
70. Zhao Y, Chen F, Wu W, et al. GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Mucosal Immunol. 2018;11:752-62.
71. Kasahara K, Krautkramer KA, Org E, et al. Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nat Microbiol. 2018;3:1461-71.
72. Zheng L, Kelly CJ, Battista KD, et al. Microbial-derived butyrate promotes epithelial barrier function through IL-10 receptor-dependent repression of claudin-2. J Immunol. 2017;199:2976-84.
73. Kaiko GE, Ryu SH, Koues OI, et al. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell. 2016;165:1708-20.
74. Kim S, Shin Y, Kim T, et al. Mucin degrader Akkermansia muciniphila accelerates intestinal stem cell-mediated epithelial development. Gut Microbes. 2021;13:1892441.
75. Parada Venegas D, De La Fuente MK, Landskron G, et al. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. 2019;10:277.
76. Singh N, Gurav A, Sivaprakasam S, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40:128-39.
77. Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569-73.
78. Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446-50.
79. Chen L, Sun M, Wu W, et al. Microbiota metabolite butyrate differentially regulates Th1 and Th17 cells’ differentiation and function in induction of colitis. Inflamm Bowel Dis. 2019;25:1450-61.
80. Park J, Kim M, Kang S, et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 2015;8:80-93.
81. Yang W, Yu T, Huang X, et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat Commun. 2020;11:4457.
82. Kim M, Qie Y, Park J, Kim CH. Gut microbial metabolites fuel host antibody responses. Cell Host Microbe. 2016;20:202-14.
83. Zhao Y, Liu J, Hao W, et al. Structure-specific effects of short-chain fatty acids on plasma cholesterol concentration in male syrian hamsters. J Agric Food Chem. 2017;65:10984-92.
84. Haghikia A, Zimmermann F, Schumann P, et al. Propionate attenuates atherosclerosis by immune-dependent regulation of intestinal cholesterol metabolism. Eur Heart J. 2022;43:518-33.
85. Tayyeb JZ, Popeijus HE, Mensink RP, Konings MCJM, Mokhtar FBA, Plat J. Short-chain fatty acids (except hexanoic acid) lower NF-kB transactivation, which rescues inflammation-induced decreased apolipoprotein A-I transcription in HepG2 cells. Int J Mol Sci. 2020;21:5088.
86. Tayyeb JZ, Popeijus HE, Mensink RP, Plat J. Butyric acid added apically to intestinal Caco-2 cells elevates hepatic ApoA-I transcription and rescues lower ApoA-I expression in inflamed HepG2 cells co-cultured in the basolateral compartment. Biomolecules. 2021;11:71.
87. Du Y, Li X, Su C, et al. Butyrate protects against high‐fat diet‐induced atherosclerosis via up‐regulating ABCA1 expression in apolipoprotein E‐deficiency mice. Br J Pharmacol. 2020;177:1754-72.
88. Woollett LA, Wang Y, Buckley DD, et al. Micellar solubilisation of cholesterol is essential for absorption in humans. Gut. 2006;55:197-204.
89. Farr S, Stankovic B, Hoffman S, et al. Bile acid treatment and FXR agonism lower postprandial lipemia in mice. Am J Physiol Gastrointest Liver Physiol. 2020;318:G682-93.
90. Roessler J, Zimmermann F, Schumann P, et al. Modulation of the serum metabolome by the short-chain fatty acid propionate: potential implications for its cholesterol-lowering effect. Nutrients. 2024;16:2368.
91. Li M, Van Esch BCAM, Henricks PAJ, Folkerts G, Garssen J. The anti-inflammatory effects of short chain fatty acids on lipopolysaccharide- or tumor necrosis factor α-stimulated endothelial cells via activation of GPR41/43 and inhibition of HDACs. Front Pharmacol. 2018;9:533.
92. Liu T, Li J, Liu Y, et al. Short-chain fatty acids suppress lipopolysaccharide-induced production of nitric oxide and proinflammatory cytokines through inhibition of NF-κB pathway in RAW264.7 cells. Inflammation. 2012;35:1676-84.
93. Wu J, Jiang Z, Zhang H, et al. Sodium butyrate attenuates diabetes-induced aortic endothelial dysfunction via P300-mediated transcriptional activation of Nrf2. Free Radic Biol Med. 2018;124:454-65.
94. González-bosch C, Boorman E, Zunszain PA, Mann GE. Short-chain fatty acids as modulators of redox signaling in health and disease. Redox Biol. 2021;47:102165.
95. Saeedi Saravi SS, Pugin B, Constancias F, et al. Gut microbiota-dependent increase in phenylacetic acid induces endothelial cell senescence during aging. Nat Aging. 2025;5:1025-45.
96. Mathew OP, Ranganna K, Mathew J, et al. Cellular effects of butyrate on vascular smooth muscle cells are mediated through disparate actions on dual targets, histone deacetylase (HDAC) activity and PI3K/Akt signaling network. Int J Mol Sci. 2019;20:2902.
97. Pewowaruk R, Jaeger BC, Hughes TM, et al. Effects of blood pressure control on arterial stiffness mechanisms in SPRINT: a randomized controlled trial. Hypertension. 2025;82:1004-11.
98. Natarajan N, Hori D, Flavahan S, et al. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol Genomics. 2016;48:826-34.
99. Kaye DM, Shihata WA, Jama HA, et al. Deficiency of prebiotic fiber and insufficient signaling through gut metabolite-sensing receptors leads to cardiovascular disease. Circulation. 2020;141:1393-403.
100. Nakai M, Ribeiro RV, Stevens BR, et al. Essential hypertension is associated with changes in gut microbial metabolic pathways: a multisite analysis of ambulatory blood pressure. Hypertension. 2021;78:804-15.
101. Dinakis E, Nakai M, Gill PA, et al. The gut microbiota and their metabolites in human arterial stiffness. Heart Lung Circ. 2021;30:1716-25.
102. Pluznick JL, Protzko RJ, Gevorgyan H, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci USA. 2013;110:4410-5.
103. Wu Y, Xu H, Tu X, Gao Z. The role of short-chain fatty acids of gut microbiota origin in hypertension. Front Microbiol. 2021;12:730809.
104. Xu J, Moore BN, Pluznick JL. Short-chain fatty acid receptors and blood pressure regulation: council on hypertension mid-career award for research excellence 2021. Hypertension. 2022;79:2127-37.
105. R. Muralitharan R, Zheng T, Dinakis E, et al. Gut microbiota metabolites sensed by host GPR41/43 protect against hypertension. Circ Res. 2025;136:e20-33.
106. Salvado R, Lugones-Sánchez C, Santos-Minguez S, et al. Sex differences in gut microbiota and their relation to arterial stiffness (MIVAS study). Nutrients. 2024;17:53.
107. Tsai H, Tsai W, Yu P, Hung W, Hung W, Tsai Y. Circulating short-chain fatty acids and subclinical cardiovascular disease in type 2 diabetes mellitus. Pol Arch Intern Med. 2025;135:17128.
109. Cai J, Rimal B, Jiang C, Chiang JY, Patterson AD. Bile acid metabolism and signaling, the microbiota, and metabolic disease. Pharmacol Ther. 2022;237:108238.
110. Collins SL, Stine JG, Bisanz JE, Okafor CD, Patterson AD. Bile acids and the gut microbiota: metabolic interactions and impacts on disease. Nat Rev Microbiol. 2022;21:236-47.
111. Zhang Z, Lv T, Wang X, et al. Role of the microbiota-gut-heart axis between bile acids and cardiovascular disease. Biomed Pharmacother. 2024;174:116567.
112. Liu T, Kern JT, Jain U, et al. Western diet induces Paneth cell defects through microbiome alterations and farnesoid X receptor and type I interferon activation. Cell Host Microbe. 2021;29:988-1001.e6.
113. Fu T, Coulter S, Yoshihara E, et al. FXR regulates intestinal cancer stem cell proliferation. Cell. 2019;176:1098-112.e18.
114. Sorrentino G, Perino A, Yildiz E, et al. Bile acids signal via TGR5 to activate intestinal stem cells and epithelial regeneration. Gastroenterology. 2020;159:956-68.e8.
115. Li T, Ding N, Guo H, et al. A gut microbiota-bile acid axis promotes intestinal homeostasis upon aspirin-mediated damage. Cell Host Microbe. 2024;32:191-208.e9.
116. Chen L, Jiao T, Liu W, et al. Hepatic cytochrome P450 8B1 and cholic acid potentiate intestinal epithelial injury in colitis by suppressing intestinal stem cell renewal. Cell Stem Cell. 2022;29:1366-81.e9.
117. Sorribas M, Jakob MO, Yilmaz B, et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J Hepatol. 2019;71:1126-40.
118. Cai J, Sun L, Gonzalez FJ. Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe. 2022;30:289-300.
119. Guo C, Xie S, Chi Z, et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity. 2016;45:802-16.
120. Yang Y, Hao C, Jiao T, et al. Osteoarthritis treatment via the GLP-1-mediated gut-joint axis targets intestinal FXR signaling. Science. 2025;388:eadt0548.
121. Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731-44.
122. Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Investig. 2004;113:1408-18.
123. Claudel T, Inoue Y, Barbier O, et al. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology. 2003;125:544-55.
124. Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147-91.
125. De Boer JF, Schonewille M, Boesjes M, et al. Intestinal farnesoid X receptor controls transintestinal cholesterol excretion in mice. Gastroenterology. 2017;152:1126-38.e6.
126. Huang F, Zheng X, Ma X, et al. Theabrownin from Pu-erh tea attenuates hypercholesterolemia via modulation of gut microbiota and bile acid metabolism. Nat Commun. 2019;10:4971.
127. Joyce SA, Macsharry J, Casey PG, et al. Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc Natl Acad Sci USA. 2014;111:7421-6.
128. Clifford BL, Sedgeman LR, Williams KJ, et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 2021;33:1671-84.e4.
129. Chan AP, Jarrett KE, Lai RW, et al. Bile acids regulate lipid metabolism through selective actions on fatty acid absorption. Cell Metab. 2026;38:263-80.e10.
130. Velazquez-villegas LA, Perino A, Lemos V, et al. TGR5 signalling promotes mitochondrial fission and beige remodelling of white adipose tissue. Nat Commun. 2018;9:245.
131. Watanabe M, Houten SM, Mataki C, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484-9.
132. Broeders EP, Nascimento EB, Havekes B, et al. The bile acid chenodeoxycholic acid increases human brown adipose tissue activity. Cell Metab. 2015;22:418-26.
133. She J, Tuerhongjiang G, Guo M, et al. Statins aggravate insulin resistance through reduced blood glucagon-like peptide-1 levels in a microbiota-dependent manner. Cell Metab. 2024;36:408-21.e5.
134. He F, Li J, Mu Y, et al. Downregulation of endothelin-1 by farnesoid X receptor in vascular endothelial cells. Circ Res. 2006;98:192-9.
135. Voiosu A, Wiese S, Voiosu T, Bendtsen F, Møller S. Bile acids and cardiovascular function in cirrhosis. Liver Int. 2017;37:1420-30.
136. Kida T, Tsubosaka Y, Hori M, Ozaki H, Murata T. Bile acid receptor TGR5 agonism induces NO production and reduces monocyte adhesion in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:1663-9.
137. Bennett BJ, Vallim TQDA, Wang Z, et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17:49-60.
138. Wahlström A, Sayin SI, Marschall H, Bäckhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016;24:41-50.
139. Lu H, Wu Z, Wan M, et al. Taurochenodeoxycholic acid alleviates obesity-induced endothelial dysfunction. Eur Heart J. 2026;47:1221-38.
140. Liu H, Tian R, Wang H, et al. Gut microbiota from coronary artery disease patients contributes to vascular dysfunction in mice by regulating bile acid metabolism and immune activation. J Transl Med. 2020;18:382.
141. Battson ML, Lee DM, Jarrell DK, et al. Tauroursodeoxycholic acid reduces arterial stiffness and improves endothelial dysfunction in type 2 diabetic mice. J Vasc Res. 2017;54:280-7.
142. Kaffe E, Roulis M, Zhao J, et al. Humanized mouse liver reveals endothelial control of essential hepatic metabolic functions. Cell. 2023;186:3793-809.e26.
143. Lichtenstein L, Cheng CW, Bajarwan M, et al. Endothelial force sensing signals to parenchymal cells to regulate bile and plasma lipids. Sci Adv. 2024;10:eadq3075.
144. Zhang P, Li X, Liang J, et al. Chenodeoxycholic acid modulates cholestatic niche through FXR/Myc/P-selectin axis in liver endothelial cells. Nat Commun. 2025;16:2093.
145. Qu Q, Chen Y, Wang Y, et al. Lithocholic acid phenocopies anti-ageing effects of calorie restriction. Nature. 2024;643:192-200.
146. Zhu Y, Dwidar M, Nemet I, et al. Two distinct gut microbial pathways contribute to meta-organismal production of phenylacetylglutamine with links to cardiovascular disease. Cell Host Microbe. 2023;31:18-32.e9.
147. Song Y, Wei H, Zhou Z, et al. Gut microbiota-dependent phenylacetylglutamine in cardiovascular disease: current knowledge and new insights. Front Med. 2024;18:31-45.
148. Poesen R, Claes K, Evenepoel P, et al. Microbiota-derived phenylacetylglutamine associates with overall mortality and cardiovascular disease in patients with CKD. J Am Soc Nephrol. 2016;27:3479-87.
149. Nemet I, Saha PP, Gupta N, et al. A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell. 2020;180:862-77.e22.
150. Li Z, Gu M, Zaparte A, et al. Alcohol-induced gut microbial reorganization and associated overproduction of phenylacetylglutamine promotes cardiovascular disease. Nat Commun. 2024;15:10788.
151. Su I, Wu T, Liu C, Lin Y, Hsu B. Serum phenylacetylglutamine is a potential risk factor for aortic stiffness in patients with chronic hemodialysis. Diagnostics. 2025;15:3123.
152. Yang H, Chen Y, Ho C, Hsu B. Serum phenylacetylglutamine among potential risk factors for arterial stiffness measuring by carotid-femoral pulse wave velocity in patients with kidney transplantation. Toxins. 2024;16:111.
153. Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57-63.
154. Xue C, Li G, Zheng Q, et al. Tryptophan metabolism in health and disease. Cell Metab. 2023;35:1304-26.
155. Suzuki K, Susaki EA, Nagaoka I. Lipopolysaccharides and cellular senescence: involvement in atherosclerosis. Int J Mol Sci. 2022;23:11148.
156. Liu W, Wang J, Yang H, et al. The metabolite indole‐3‐acetic acid of Bacteroides Ovatus improves atherosclerosis by restoring the polarisation balance of M1/M2 macrophages and inhibiting inflammation. Adv Sci. 2025;12:2413010.
157. Alsulami M, Alamri H, Barhoumi T, Munawar N, Alghanem B. The effect of TMAO on aging-associated cardiovascular and metabolic pathways and emerging therapies. Mol Cell Biochem. 2025;480:5659-69.
158. Xue H, Chen X, Yu C, et al. Gut microbially produced indole-3-propionic acid inhibits atherosclerosis by promoting reverse cholesterol transport and its deficiency is causally related to atherosclerotic cardiovascular disease. Circ Res. 2022;131:404-20.
159. Zhu W, Gregory JC, Org E, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165:111-24.
160. Brunt VE, Gioscia-ryan RA, Casso AG, et al. Trimethylamine-N-oxide promotes age-related vascular oxidative stress and endothelial dysfunction in mice and healthy humans. Hypertension. 2020;76:101-12.
161. Lu Y, Yang W, Qi Z, et al. Gut microbe-derived metabolite indole-3-carboxaldehyde alleviates atherosclerosis. Sig Transduct Target Ther. 2023;8:378.
162. Merk D, Cox FF, Jakobs P, Prömel S, Altschmied J, Haendeler J. Dose-dependent effects of lipopolysaccharide on the endothelium—sepsis versus metabolic endotoxemia-induced cellular senescence. Antioxidants. 2024;13:443.
163. Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. 2020;78:1233-61.
164. Ecton KE, Graham EL, Risk BD, et al. Toll-like receptor 4 deletion partially protects mice from high fat diet-induced arterial stiffness despite perturbation to the gut microbiota. Front Microbiomes. 2023;2:1095997.
165. Sakura T, Morioka T, Shioi A, et al. Lipopolysaccharide-binding protein is associated with arterial stiffness in patients with type 2 diabetes: a cross-sectional study. Cardiovasc Diabetol. 2017;16:62.
166. Brunt VE, Casso AG, Gioscia-Ryan RA, et al. Gut microbiome-derived metabolite trimethylamine N-oxide induces aortic stiffening and increases systolic blood pressure with aging in mice and humans. Hypertension. 2021;78:499-511.
167. Huang P, Lin Y, Chen Y, et al. The association between serum trimethylamine N-oxide and arterial stiffness in chronic peritoneal dialysis patients: a cross-sectional study. Toxins. 2024;16:523.
168. Huang P, Hsu B, Lai Y, Wang C, Tsai J. Serum trimethylamine N-Oxide level is positively associated with aortic stiffness measured by carotid-femoral pulse wave velocity in patients undergoing maintenance hemodialysis. Toxins. 2023;15:572.
169. Hsu B, Wang C, Lin Y, Lai Y, Tsai J. Serum trimethylamine N-oxide level is associated with peripheral arterial stiffness in advanced non-dialysis chronic kidney disease patients. Toxins. 2022;14:526.
170. Wang Q, Lv H, Ainiwan M, et al. Untargeted metabolomics identifies indole-3-propionic acid to relieve Ang II-induced endothelial dysfunction in aortic dissection. Mol Cell Biochem. 2024;479:1767-86.
171. Shaheen N, Miao J, Li D, et al. Indole-3-acetic acid protects against lipopolysaccharide-induced endothelial cell dysfunction and lung injury through the activation of USP40. Am J Respir Cell Mol Biol. 2024;71:307-17.
172. Jia Y, He M, Wang F, et al. Indole-3-lactic acid protects the gut vascular barrier following intestinal ischemia injury through AhR/Nrf2/STAT3 mediated claudin 2 downregulation. Cell Commun Signal. 2025;23:447.
174. Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of vascular aging. Circ Res. 2018;123:849-67.
175. Jie Z, Xia H, Zhong S, et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun. 2017;8:845.
176. Hua R, Ding N, Hua Y, et al. Ligilactobacillus murinus and lactobacillus johnsonii suppress macrophage pyroptosis in atherosclerosis through butyrate‐GPR109A‐GSDMD axis. Adv Sci. 2025;12:e01707.
177. Qi Z, Zhang W, Zhang P, et al. The gut microbiota-bile acid-TGR5 axis orchestrates platelet activation and atherothrombosis. Nat Cardiovasc Res. 2025;4:584-601.
178. Li B, Fan H, Wu H, et al. Targeting FXR in hepatocytes: a promising approach to enhance fibrinolysis and reduce deep vein thrombosis risk. Blood. 2025;146:2464-78.
179. Hutchison ER, Mallikarjun J, Byun JH, et al. GPR41 deficiency alters the gut microbiota-bile acid axis, reduces ileal expression of Npc1l1, and attenuates hypercholesterolemia in male mice. Gut Microbes. 2025;17:2598957.
180. Mian O, Santi N, Boodhwani M, et al. Arterial age and early vascular aging, but not chronological age, are associated with faster thoracic aortic aneurysm growth. J Am Heart Assoc. 2023;12:e029466.
181. Quintana RA, Taylor WR. Cellular mechanisms of aortic aneurysm formation. Circ Res. 2019;124:607-18.
182. Song T, Zhao S, Luo S, et al. SLC44A2 regulates vascular smooth muscle cell phenotypic switching and aortic aneurysm. J Clin Investig. 2024;134:e173690.
183. Tian Z, Zhang Y, Zheng Z, et al. Gut microbiome dysbiosis contributes to abdominal aortic aneurysm by promoting neutrophil extracellular trap formation. Cell Host Microbe. 2022;30:1450-63.e8.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.