


The application of GLP-1 receptor agonists in obesity due to MC4R deficiency
 0
0 
Abstract
Although the pathogenesis of genetic obesity is becoming increasingly clear, effective treatments remain lacking, particularly for obesity caused by melanocortin 4 receptor (MC4R) deficiency. The activity of MC4R is strictly regulated by its endogenous ligands: α-melanocyte-stimulating hormone (α-MSH) binds to MC4R and transmits anorexigenic and energy-expenditure signals, whereas agouti-related protein (AgRP) exerts an antagonistic effect by binding to the receptor, competitively inhibiting α-MSH action and promoting appetite. Current options, such as lifestyle modifications and bariatric surgery, have limited efficacy, highlighting the need for accessible pharmacological interventions. This review evaluates glucagon-like peptide-1 (GLP-1) receptor agonists in MC4R-deficient obesity. We conducted a comprehensive literature search across databases, including PubMed and Web of Science, compiling data from clinical trials, case reports, and basic research studies published up to 2025, with a focus on the efficacy, safety, and mechanism of action of drugs such as liraglutide, semaglutide, and tirzepatide in patients with MC4R deficiency. Evidence indicates these agents induce significant weight loss, appetite suppression, and metabolic improvements via mechanisms independent of intact MC4R signaling. Their weight-loss effects are comparable to those seen in common obesity, with additional benefits for comorbidities like insulin resistance and dyslipidemia. These findings support the GLP-1 pathway as an alternative to impaired MC4R, offering a viable treatment. Understanding their efficacy and mechanisms provides new treatment options for this rare condition and offers evidence for genetically specific personalized weight management.

Keywords
INTRODUCTION
Genetic obesity is broadly divided into syndromic obesity and non-syndromic obesity, both of which can be caused by monogenic, polygenic, or chromosomal abnormalities[1]. According to the latest genetic research, more refined genetic risk stratification is now complementing traditional etiological classification. A groundbreaking study conducted by Chami et al., using large-scale whole-genome analysis, revealed that obesity has a polygenic basis and identified at least eight genetic subtypes with distinct profiles of complication risk. These subtypes are defined by 266 unique uncoupling genetic variants, which increase obesity risk while simultaneously reducing cardiovascular and metabolic risks. This discovery provides a novel mechanistic perspective for understanding obesity and its related diseases[2]. In addition to its low incidence rate, genetic obesity also differs significantly from common obesity in its clinical manifestations. It is typically characterized by early onset (in childhood and adolescence), more pronounced weight gain, higher body fat percentage, and more severe hyperphagia, often accompanied by endocrine disorders (such as hypogonadism). Patients with syndromic obesity also exhibit congenital malformations and intellectual disabilities. These features are important clues for the early clinical identification and diagnosis of genetic obesity[3]. With the development of genetic testing technologies such as genome-wide association studies (GWAS) and next-generation sequencing (NGS), it has been discovered that approximately 127 locus abnormalities in the human genome are associated with the onset and progression of obesity[4], as along with more than 550 monogenic mutations related to obesity[5].
In patients with severe early-onset obesity, most of the common mutations are clustered in the leptin-melanocortin pathway (LMP), which mainly regulates appetite to control the body’s energy balance. In this pathway, adipocytes secrete leptin, which transmits energy storage signals to the hypothalamus. Through a cascade of reactions, this process ultimately generates signals that suppress appetite and increase energy consumption. As illustrated in Figure 1, adipocyte-derived leptin signals peripheral energy status to the arcuate nucleus (ARC) of the hypothalamus. Within the ARC, two primary, functionally antagonistic neuronal populations serve as first-order neurons: the anorexigenic pro-opiomelanocortin (POMC)-expressing neurons and the orexigenic agouti-related protein (AgRP)/neuropeptide Y (NPY)-expressing neurons[6].
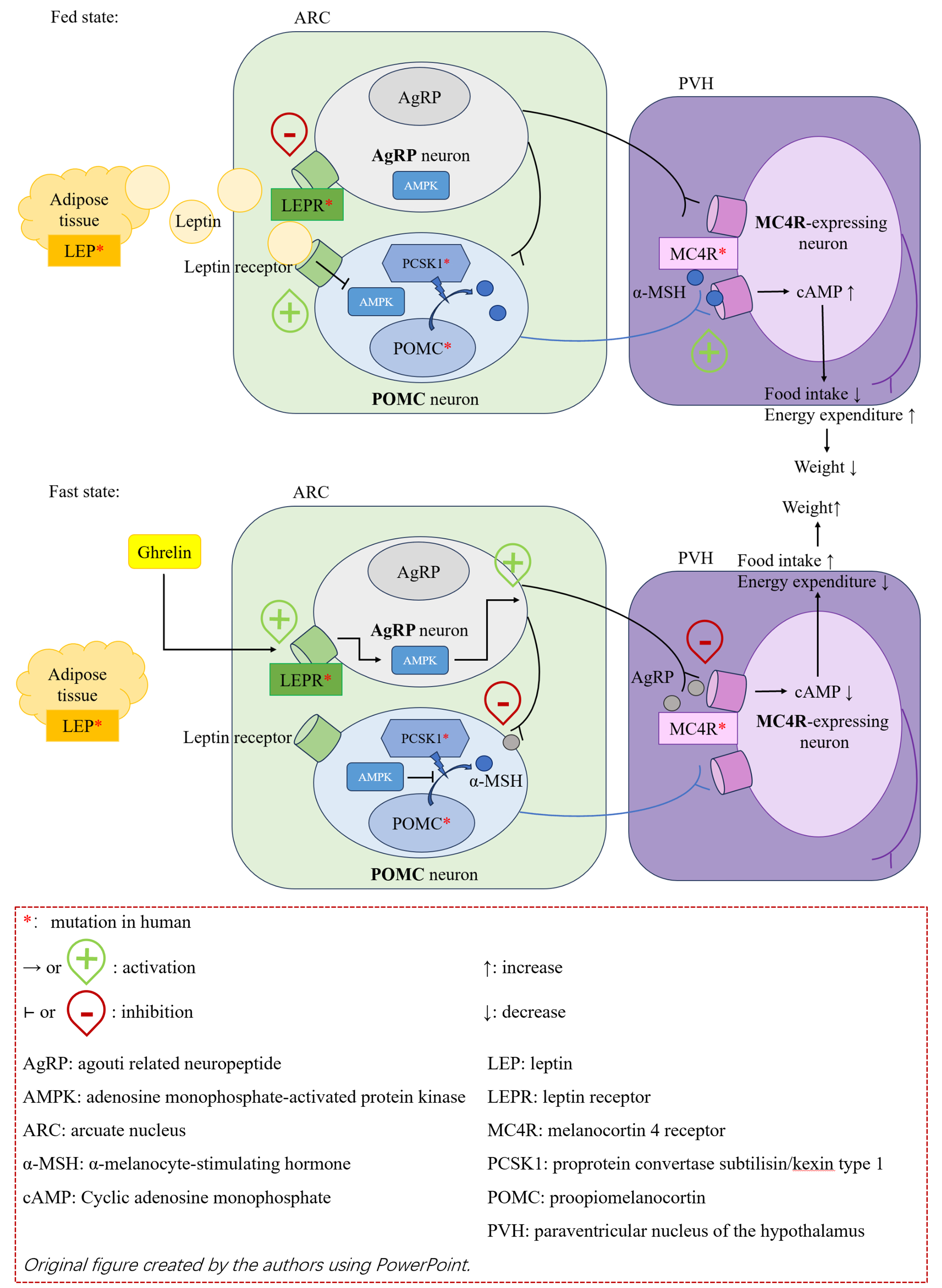
Figure 1. Leptin-melanocortin pathway.
In the fed state, elevated leptin levels activate POMC neurons. This activation is not merely a linear switch but also dynamically regulated by intracellular energy sensors. For instance, leptin suppresses the activity of AMP-activated protein kinase (AMPK) in POMC neurons, which is permissive for their firing and subsequent peptide production[7]. Activated POMC neurons process the precursor polypeptide POMC via the action of POMC by proprotein convertase subtilisin/kexin type 1 (PCSK1) to generate, among other peptides, α-melanocyte-stimulating hormone (α-MSH)[8]. α-MSH acts as the principal endogenous agonist for the melanocortin 4 receptor (MC4R), which is highly expressed on second-order neurons in the paraventricular nucleus (PVN) of the hypothalamus. Binding of α-MSH to MC4R triggers a Gs protein-coupled signaling cascade, which increases intracellular cyclic AMP (cAMP) levels and ultimately activates downstream pathways that promote satiety and increase energy expenditure[9].
Conversely, in the fasted or low-energy state, falling leptin levels relieve the tonic inhibition on AgRP/NPY neurons while simultaneously failing to activate POMC neurons. The activation state of AgRP neurons is also tightly controlled by metabolic cues; for example, the orexigenic hormone ghrelin activates these neurons in part by modulating their AMPK activity[7]. Activated AgRP neurons release two key neurotransmitters to promote feeding: (1) NPY, which acts on Y receptors to potently stimulate food intake; and (2) AgRP, which functions as an inverse agonist at MC4R. AgRP not only competitively blocks the binding of α-MSH but also constitutively suppresses the basal activity of MC4R, thereby antagonizing the anorexigenic signaling[10]. Furthermore, AgRP neurons send potent inhibitory projections [via gamma-aminobutyric acid (GABA)] directly onto POMC neurons, providing a second, parallel mechanism to suppress satiety signaling and promote a robust orexigenic drive[11]. The integrated output from the PVN and other hypothalamic nuclei ultimately dictates the net behavioral and metabolic response.
As shown in Table 1[1,8,12-19], because of the critical role of this pathway in energy regulation, functional abnormalities caused by mutations in any of its critical genes (e.g., LEP, LEPR, POMC, PCSK1, MC4R) can lead to severe early-onset obesity and extreme hyperphagia and are often accompanied by endocrine abnormalities due to deficiencies in related hormones, such as hypogonadism and hypothyroidism[12,20]. Beyond the core signaling molecules, transcriptional regulation and neurodevelopment within this pathway are equally crucial. For instance, the pleckstrin homology domain interacting protein (PHIP) protein acts as a transcriptional co-regulator that directly modulates POMC transcription, whereas the SIM1 transcription factor is indispensable for the normal development of the PVN and the proper expression of MC4R[8,13]. Mutations in these genes not only cause severe obesity but also frequently lead to a broad spectrum of neurological phenotypes, including intellectual disability, developmental delay, and behavioral abnormalities (e.g., autism-like features), highlighting the foundational role of hypothalamic development in energy homeostasis[12,14]. Furthermore, adaptor proteins and neurotrophic factor receptors located downstream of leptin and MC4R signaling are also critical. SH2B1, as a downstream signaling molecule of both LEPR and the insulin receptor, exacerbates hyperphagia and insulin resistance when deficient[15]. Mutations in the neurotrophic factor receptor neurotrophic receptor tyrosine kinase 2 (NTRK2), which affect neuronal survival and synaptic plasticity, contribute to a complex array of phenotypes including obesity, severe intellectual disability, and blunted pain response[8,12]. In addition, mutations in genes related to other functions - such as ciliary function (e.g., BBS1-20, ALMS1), adipocyte function and energy expenditure (e.g., PPARG, CEP19/KSR2) - are also involved in the pathogenesis of hereditary obesity[7]. These findings reveal the high degree of molecular heterogeneity underlying genetic obesity and indicate that its clinical manifestations can range from relatively isolated severe metabolic disturbances to complex syndromes accompanied by significant neurodevelopmental defects.
Important gene mutation types in monogenic obesity
| Gene | Function | Phenotype |
| LEP | Adipocyte-derived hormone; signals peripheral energy stores to the hypothalamus[1] | Severe early-onset obesity, severe hyperphagia[1], hypogonadotropic hypogonadism, hypothyroidism, impaired immune function[12] |
| LEPR | Leptin receptor[1] | Similar to that of LEP deficiency[12] |
| POMC | Precursor of α-MSH and ACTH[8] | Severe obesity, ACTH-deficient adrenal insufficiency, mild hypothyroidism, and red hair with fair skin (variable by ethnicity)[12] |
| PCSK1 | Prohormone convertase; processes POMC and other precursors[16] | Severe obesity, multiple endocrine disorders (e.g., hypothyroidism, diabetes mellitus), intestinal malabsorptive diarrhea, and central diabetes insipidus[17,18] |
| MC4R | Receptor for α-MSH; key regulator of energy balance[19] | Hyperphagia, accelerated linear growth, and hyperinsulinemia[19] |
| PHIP | Transcriptional coregulator of POMC[8] | Severe obesity, developmental retardation, intellectual disability, behavioral disorders (e.g., emotional lability, autism-like behaviors), hypotonia, and distinctive facial features[14] |
| SH2B1 | Adaptor protein downstream of LEPR and insulin receptor[15] | Severe obesity, hyperphagia, insulin resistance, and behavioral disorders[15] |
| NTRK2 | Brain-derived neurotrophic factor homologous receptors located downstream of MC4R; regulate neuronal development and survival[8] | Severe obesity, developmental retardation, intellectual disability, impaired memory, behavioral disorders, and blunted pain response[12] |
| SIM1 | Transcription factor; regulates PVN development and MC4R expression[13] | Severe obesity, behavioral disorders[12] |
PATHOGENIC MECHANISMS AND CLINICAL MANIFESTATIONS OF MC4R-DEFICIENT OBESITY
Numerous genetic screening studies in cohorts with severe early-onset obesity have confirmed that obesity due to MC4R deficiency is currently the most common cause of monogenic obesity, highlighting the critical role of MC4R in energy balance regulation[21-24]. The human MC4R gene is located on chromosome 18q21.32. The MC4R protein belongs to the Class A G protein-coupled receptor family, consists of 332 amino acids, and has seven transmembrane domains. It is mainly expressed in the PVN of the hypothalamus (PVH), the spinal cord, and sympathetic neurons. To date, over 150 pathogenic variants have been identified in the MC4R gene, including missense, nonsense, and frameshift mutations. These variants are widely distributed across both intracellular and extracellular domains of the receptor, leading to loss-of-function (LOF) or hypofunction through diverse molecular mechanisms, as summarized in Table 2[25-28].
Common types of MC4R dysfunction
| Impaired functions | Mutation examples | Molecular mechanism |
| Protein synthesis | p.Tyr35Stop | Truncated protein or mRNA degradation; complete loss of functional MC4R[25] |
| Intracellular transportation | p.Asp90Asn, p.Ile125Lys | The MC4R precursor protein misfolds in the endoplasmic reticulum and is ultimately degraded intracellularly, and fails to traffic to the cell membrane surface[26] |
| Ligand binding | p.Asp126Tyr | Normal membrane expression; reduced α-MSH binding affinity[27] |
| Signal transduction | p.Arg165Gln | Normal expression and binding; impaired Gs-cAMP activation[28] |
Many variants (e.g., Ser127Leu) exhibit multiple concurrent functional defects, and the majority follow a dominant inheritance pattern consistent with haploinsufficiency[29].
Heterozygous MC4R variants are not only the most common monogenic cause of severe obesity in children but also a frequent cause of severe late-onset obesity in adults. The loss or impairment of MC4R function compromises the generation of satiety signals, which results in persistently elevated appetite and ultimately drives the development of severe obesity. Notably, despite the presence of severe obesity, large-scale population cohort studies have revealed that individuals with MC4R-deficient obesity exhibit a unique metabolically protective phenotype. Compared to individuals with obesity of other etiologies matched for BMI (body mass index), they have lower levels of total cholesterol, low-density lipoprotein cholesterol (LDL-C), and triglycerides. Moreover, the risks of coronary heart disease, myocardial infarction, and hypertension are significantly attenuated, even approaching those of normal-weight individuals. This distinctive phenotype refines the urgency and goals of weight management. In contrast to common obesity, where cardiovascular risk reduction drives intervention, the main rationale for treatment in MC4R-deficient patients shifts toward alleviating mechanical complications (e.g., sleep apnea, osteoarthritis), preventing diabetes (hyperinsulinemia is common despite relative euglycemia), and improving quality of life[30]. Furthermore, whether the cardiovascular benefits of GLP-1 receptor agonists (GLP-1 RAs) apply to to this population requires nuanced interpretation. While agents such as semaglutide have proven cardiovascular protective effects in large outcome trials[31], these studies predominantly enrolled patients with established cardiovascular disease or high-risk profiles. Whether the same magnitude of risk reduction occurs in a population with inherited cardioprotection remains unknown, as the low baseline risk may attenuate absolute benefits - a phenomenon known as a ceiling effect. However, the direct vascular protective effects of GLP-1 RAs, including anti-inflammatory and endothelial actions, could theoretically operate independently of baseline metabolic status[31]. Most importantly, cardiovascular monitoring remains warranted despite favorable population-level profiles, as long-standing severe obesity can itself affect cardiac structure and function, and individual-level events are not precluded. Finally, this unique phenotype offers mechanistic insights: the dissociation between severe adiposity and a favorable cardiometabolic profile suggests that MC4R signaling plays a non-redundant role in integrating energy balance with cardiovascular regulation[2,32,33]. For GLP-1 RA therapy, this implies that weight loss in this population likely derives primarily from appetite suppression rather than from correcting pre-existing metabolic abnormalities, informing expectations about therapeutic mechanisms and outcomes.
Although there is still a lack of expert consensus and guidelines for the diagnosis of MC4R-deficient obesity are currently lacking, preliminary screening for suspected MC4R variant carriers can be accomplished by accurate identification of patients’ typical clinical features. Then, through standardized genetic testing techniques and the American College of Medical Genetics and Genomics (ACMG) classification criteria for genetic disease testing, a comprehensive diagnosis can be made for early identification of obesity due to MC4R deficiency. Common clinical manifestations of MC4R variants are summarized in Table 3[23,34-38]; patients with these features should be strongly suspected of having MC4R-deficient obesity, and genetic evaluation should be considered.
Common clinical manifestations of MC4R variants
| Feature | Manifestation |
| Age of onset | Early, typically occurring between 1 and 5 years of age, or even in infancy[34] |
| Appetite | Severe and persistent hyperphagia, often manifested as unquenchable hunger, an intense preoccupation with food, and behaviors such as food stealing or hoarding[23,34] |
| Level of obesity | Severe obesity (BMI ≥ 37.5 kg/m2): Weight-for-height or BMI is consistently well above the 99th percentile or higher for age and sex[35] |
| Linear growth | Accelerated growth in height and bone age, with height often above the average level for peers and significantly increased bone mineral density[23,34] |
| Endocrine and metabolic abnormalities | Early-onset hyperinsulinemia, which may be accompanied by insulin resistance, though severe hyperglycemia is relatively rare[23,36]; hyperlipidemia, such as hypertriglyceridemia and hypercholesterolemia[37,38] |
| Associated symptoms | Isolated obesity, generally not accompanied by intellectual disability, organ malformations, or distinctive facial features[23,34] |
PHARMACOLOGICAL THERAPIES FOR MC4R-DEFICIENT OBESITY
While lifestyle modifications such as dietary restriction and physical activity provide only modest weight loss benefits, large-scale cohort studies have demonstrated that strict conventional interventions often result in suboptimal weight reduction outcomes among MC4R variant carriers[39-42].
Bariatric surgery is widely recognized as the most effective intervention for severe obesity, yielding significant and sustained weight loss in most patients. However, its efficacy is substantially compromised in patients with obesity caused by MC4R deficiency, a common monogenic form of obesity. Accumulating evidence from clinical studies, including case-control studies, systematic reviews, and meta-analyses, has consistently demonstrated the limited long-term weight loss efficacy of bariatric surgery in MC4R-deficient patients, which differs significantly from its effects in patients with obesity induced by other genetic variants or non-genetic factors.
A consistent finding across relevant studies is that bariatric surgery achieves comparable short-term weight loss in MC4R-deficient patients and non-deficient controls; however, this initial advantage diminishes over time, with significantly poorer long-term outcomes as patients tend to regain much of the weight. In a case-control study involving 105 patients[43], heterozygous MC4R pathogenic/likely pathogenic (P/LP) variant carriers exhibited similar maximum total body weight loss percentage (%TBWLmax) to non-carriers (28.3% ± 8.5% vs. 28.7% ± 7.8%), but they experienced more severe long-term weight regain (median regain: 14.2 kg vs. 9.8 kg) and poorer final weight loss outcomes (final %TBWL: 18.5% ± 10.2% vs. 23.1% ± 9.5%). Notably, variants of uncertain significance in MC4R did not exert a significant impact on long-term surgical outcomes, indicating that only P/LP variants are associated with compromised long-term efficacy. Similar results were observed in a study focusing on MC4R deficiency and pharmacotherapy[44], where MC4R-deficient patients who underwent sleeve gastrectomy achieved stable short-term weight loss (mean 17.99% weight loss within 6 months), but long-term outcomes were heterogeneous and often accompanied by weight regain, requiring revisional surgery in some cases. The underlying mechanism for this phenomenon is presumably the persistent dysregulation of energy homeostasis caused by MC4R deficiency, which cannot be fully reversed by bariatric surgery alone, leading to excessive food intake and subsequent weight regain over time.
MC4R is a key component of the LMP, which plays a crucial role in regulating appetite and energy metabolism. Mutations in other genes of this pathway (e.g., LEPR, POMC) also cause monogenic obesity, but the efficacy of bariatric surgery in these patients differs from that in MC4R-deficient patients. For patients with bi-allelic mutations in POMC, LEPR, or MC4R[45], bariatric surgery achieved only limited long-term weight loss, with all patients experiencing significant weight regain after initial weight reduction. The median maximum excess weight loss percentage (%EWLmax) was 47.5%, but the final median %EWL decreased to only 24.2% after long-term follow-up (up to 19 years). Notably, bi-allelic MC4R mutation carriers had long-term outcomes as poor as those with bi-allelic LEPR or POMC mutations, suggesting that biallelic mutations in LMP genes generally lead to more severe impairment of surgical efficacy compared to heterozygous mutations. In contrast, a 15-year case-control study[46] evaluated the outcomes of Roux-en-Y gastric bypass in patients with heterozygous LMP variants (including MC4R, LEPR, POMC, and PCSK1). The study found that heterozygous LMP variant carriers had a significantly higher weight regain percentage (52.7% ± 29.7% vs. 29.8% ± 20.7%) and poorer final %TBWL (16.6% ± 10.7% vs. 28.7%) compared to non-carriers at 15 years of follow-up. Although the study did not distinguish the efficacy among different heterozygous LMP variants, it confirmed that heterozygous mutations in the LMP pathway (including MC4R) are associated with compromised long-term surgical outcomes, consistent with the findings in heterozygous MC4R P/LP variant carriers. A systematic review and meta-analysis[47] further quantified the impact of LMP mutations on bariatric surgery outcomes. The results indicated that LMP mutation carriers had a 3.03% lower overall weight loss compared to non-carriers, with no significant difference in short-term outcomes (≤ 6 months) but a significant reduction in long-term outcomes. Subgroup analysis indicated that homozygous mutations were associated with more severe impairment of long-term weight loss than heterozygous mutations, which explains why bi-allelic MC4R mutation carriers have poorer long-term outcomes than heterozygous carriers.
In conclusion, the evidence from multiple studies confirms that the long-term efficacy of bariatric surgery in MC4R-deficient obesity is limited, which is closely related to the type of MC4R mutation (homozygous vs. heterozygous) and consistent with the impact of other LMP pathway mutations on surgical outcomes. Therefore, regarding surgical indications, the available evidence argues against a one-size-fits-all approach. Surgery should not be categorically contraindicated in MC4R-deficient patients, but careful patient selection and informed consent are paramount.
Regarding pharmacological therapy, no medications are currently specifically indicated for MC4R variants are currently available. However, several drugs have been approved and clinically implemented for obesity caused by other genetic mutations in the MC4R pathway, as summarized in Table 4.
Clinical studies on pharmacological therapies for MC4R-deficient obesity
| Reference | Drug (s) | Study type | Evidence level | Sample (MC4R Pts) | Key findings |
| Collet et al., 2017[55] | Setmelanotide | Phase Ib RCT | III | n = 8 (het LOF) | Weight loss (-3.5 kg) and waist reduction (-5.8 cm) in 28 days. No CV side effects |
| Iepsen et al., 2018[67] | Liraglutide | Clinical study | IV | n = 4 (het) | ~6% weight loss in 16 weeks. Reduced hunger and energy intake by 28% |
| Iepsen et al., 2020[68] | Liraglutide | Case report | V | n = 1 (hom LOF) | Weight loss (-9.7 kg) in 16 weeks. Improved glycemia and lipids |
| Iepsen et al., 2020[70] | Liraglutide | Observational cohort | IV | n = 17 (het) | ~6% weight loss. No bone density benefit seen (unlike controls) |
| Zaitoon et al., 2023[69] | Liraglutide + Metformin | Case report | V | n = 1 (hom LOF, child) | Halted weight gain in 1 year. Improved metabolism, behavior and mood |
| Gokul et al., 2025[76] | Semaglutide | Case report | V | n = 1 (het, child) | Major weight (-20.6 kg) and body fat reduction in 1 year. Metabolic and QoL improvement |
| Gad et al., 2024[77] | Semaglutide | Case report | V | n = 2 (siblings, het) | Mixed weight response. Showed neuroprotective effect (nerve fiber regeneration) |
| Bhatnagar et al., 2025[87] (SURMOUNT-1) | Tirzepatide | Post-hoc subgroup analysis of RCT | II-III | n = 32 (carriers) | 18.3% weight loss in 72 weeks, equal to non-carriers. Metabolic benefits are maintained |
Before reviewing individual pharmacological agents, it is essential to acknowledge the inherent limitations of the evidence supporting their use, specifically in MC4R-deficient obesity. As delineated in Table 4, the current literature consists predominantly of low-level evidence (Level IV and V), including case reports, small observational cohorts, and post hoc subgroup analyses of larger trials. Notably, no randomized controlled trials (RCTs) prospectively designed and adequately powered to evaluate outcomes specifically in MC4R-deficient populations have been completed to date. No systematic data exist regarding developmental safety, effects on linear growth and pubertal progression, or long-term cardiometabolic consequences in this population.
This evidentiary constraint necessitates particular caution when extrapolating efficacy and safety findings from common obesity to MC4R-deficient obesity. While the clinical data reviewed herein provide a compelling rationale for the off-label application of these agents in MC4R-deficient obesity, they should be interpreted as hypothesis-generating rather than definitive. Dedicated, prospective studies with adequate sample sizes, long-term follow-up, and comprehensive metabolic phenotyping are urgently required to validate these preliminary observations and establish evidence-based, genotype-specific treatment approaches.
Given the current lack of robust evidence from long-term, large-sample RCTs, the majority of evidence for pharmacological interventions targeting MC4R-deficient obesity discussed in this review - particularly for liraglutide and semaglutide - derives from case reports. Such reports are only able to generate hypotheses and cannot establish definitive proof of therapeutic efficacy.
MC4R AGONISTS
Setmelanotide
Setmelanotide is the only drug approved by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) to treat inherited obesity. It isn’t specifically approved for MC4R deficiency specifically, only for weight management and appetite control in adults and children with POMC-, PCSK1-, or LEPR-deficient obesity or those with genetically confirmed pathogenic variants, likely pathogenic variants, or variants of uncertain significance in POMC, PCSK1, or LEPR (FDA: 2 years of age and older; EMA: 2 years of age and older)[48,49]. Setmelanotide is an 8-amino acid cyclic peptide analog of α-MSH that promotes weight loss by activating MC4R to reduce hunger, thereby decreasing caloric intake and increasing energy expenditure[50]. According to the results of the RCT for patients with monogenic obesity (LEPR or POMC deficiency) and acquired hypothalamic obesity, setmelanotide can reduce body weight and inhibit appetite to a certain extent, and common adverse reactions include injection site reactions, skin hyperpigmentation, nausea, diarrhea, and abdominal pain, with occasional occurrences of sexual adverse reactions, depression, and suicidal ideation[51,52].
Preliminary observations from a Phase III clinical trial (NCT02896192) suggested that among 10 patients with POMC-deficient obesity (aged ≥ 6 years, BMI ≥ 30 kg/m2), approximately 80% achieved a weight reduction of ≥ 10% after 1 year of treatment with setmelanotide following individualized dose titration. The data indicated that the overall mean body weight of patients was significantly reduced by 25.6% compared to baseline, while the hunger score also decreased significantly by 27.1%[53]. Given these reported outcomes regarding weight reduction and appetite control in obesity associated with the MC4R pathway, it is plausible to hypothesize that setmelanotide may exert similar effects in MC4R-deficient obesity. Supporting this possibility, preclinical studies have reported that setmelanotide can effectively restore MC4R activity in obese animal models harboring LOF MC4R variants, thereby reducing food intake and increasing total energy expenditure, which results in significant weight loss. These studies also noted that no adverse cardiovascular effects were observed[54]. Further evidence comes from a randomized, double-blind, placebo-controlled Phase Ib clinical trial that also evaluated the efficacy and safety of setmelanotide in obese patients harboring heterozygous LOF variants in MC4R (either complete or partial LOF; n = 8, with 6 patients receiving setmelanotide and 2 assigned to placebo). Results from this trial suggested that after 28 days of treatment, the setmelanotide group demonstrated a significant mean weight reduction of 3.48 kg from baseline, accompanied by a significant decrease in mean waist circumference of 5.83 cm. It should be noted that the between-group statistical difference in weight change was not statistically significant compared with the placebo group; nevertheless, the results, including weight and waist circumference reductions from baseline, provide preliminary evidence of biological activity. However, these findings are based on a very small pilot trial and require confirmation in larger, adequately powered studies. Furthermore, 24-h ambulatory monitoring indicated that setmelanotide did not induce increases in heart rate or blood pressure, pointing to favorable cardiovascular safety[55].
At present, the lack of large-scale, confirmatory clinical trials specifically targeting MC4R deficiency is largely due to significant scientific and clinical heterogeneity, which has made conducting such trials logistically challenging and commercially less appealing compared to pursuing broader or more clearly defined patient populations.
From a scientific standpoint, MC4R deficiency presents a formidable challenge for treatment with MC4R agonists like setmelanotide due to extreme genetic and functional heterogeneity. Different MC4R variants differently impact on receptor signaling, making it difficult to predict which patients would respond to agonist therapy. Setmelanotide can bypass upstream defects but cannot repair the receptor itself. In cases of complete LOF mutations in MC4R, the receptor becomes nonfunctional, rendering agonists ineffective. This directly complicates patient screening and enrollment, leads to substantially lower overall expected efficacy compared with patients harboring upstream genetic defects (such as POMC deficiency)[56], and results in a low statistical success rate in large-scale trials. The pioneering 2017 Phase 1b trial of setmelanotide (mentioned above), while providing a critical proof-of-concept for weight loss in a small cohort of heterozygous carriers, was primarily designed as an exploratory study. The trial itself explicitly concluded that while the drug showed promise, further studies were necessary to establish whether setmelanotide could elicit clinically meaningful weight loss in a subset of the MC4R-deficient obese population. This underscores the key issue: a trial powered to identify and confirm efficacy in specific, responsive genetic subsets would be highly complex and require substantial patient numbers to stratify by mutation type. This scientific uncertainty directly impacts commercial interest, as developing a drug for a fragmented and unpredictable subpopulation within an already rare disease is a high-risk investment.
Consequently, the commercial and regulatory strategy for MC4R-targeted therapies has logically shifted toward indications with more predictable and homogeneous patient populations. Rhythm Pharmaceuticals, the developer of setmelanotide, has successfully pursued regulatory approval for the drug (marketed as IMCIVREE®) in conditions caused by upstream defects in the MC4R pathway, such as POMC and LEPR deficiencies, where the receptor itself is intact, and the agonist mechanism is highly effective[57]. Furthermore, the company’s development pipeline, including a large Phase 2 basket trial enrolling patients with various rare genetic disorders of obesity, incorporates MC4R-deficient patients as just one of several cohorts[58]. This approach of targeting the entire MC4R pathway, rather than the MC4R gene alone, enables a more viable commercial model by consolidating multiple ultra-rare diseases into a single development program. This is supported by the company’s focus on expanding its MC4R franchise into adjacent areas, such as acquired hypothalamic obesity to drive growth[59].
GLP-1-BASED THERAPY
Preliminary studies suggest potential weight-loss benefits of setmelanotide in MC4R deficiency. However, the fundamental constraint remains: as an agonist of the impaired receptor itself, its mechanism is intrinsically compromised, which may limit efficacy and necessitate robust validation. This scientific uncertainty, compounded by the drug’s unavailability in China, highlights an imperative to explore alternative therapeutic pathways independent of the leptin-melanocortin system. Given this dilemma, shifting focus to alternative weight loss therapies that do not rely on the leptin-melanocortin system - such as GLP-1 RAs, which have distinctly different mechanisms of action - may offer theoretical feasibility for the treatment of MC4R-deficient obesity. Peripherally, they primarily act on the pancreas and gastrointestinal tract, stimulating insulin secretion and inhibiting glucagon secretion when blood glucose levels rise, thereby exerting a hypoglycemic effect[60]. Concurrently, they significantly delay gastric emptying and reduce caloric intake by enhancing satiety[61]. In the central nervous system, GLP-1 RAs can either directly cross regions with a compromised blood-brain barrier or act via vagal afferent signaling to target GLP-1 receptors (GLP-1Rs) in the nucleus tractus solitarius (NTS) of the brainstem. This enhances GABAergic signaling, thereby inhibiting adjacent orexigenic NPY neurons[62]. Additionally, GLP-1 RAs can modulate appetite-regulating centers in the hypothalamus, such as directly activating POMC neurons[63] or inhibiting AgRP neurons by acting on GLP-1R-expressing neurons in the dorsomedial hypothalamus[61,63]. GLP-1 RAs bypass genetic defects in the leptin-melanocortin system, promote weight loss, and improve metabolic function through the coordinated regulation of blood glucose and appetite[64,65] [Figure 2]. However, the relevance of GLP-1 signaling in genetic obesity may extend beyond this bypass mechanism. A recent study by Eckert et al. demonstrated that pharmacological activation of GLP-1 signaling ameliorated cystogenesis and renal dysfunction in a zebrafish model of nephronophthisis, a ciliopathy characterized by progressive kidney failure[66]. This finding is particularly noteworthy given that ciliary dysfunction resulting from mutations in genes such as those in the BBS family or ALMS1 represents an established mechanism underlying syndromic forms of hereditary obesity. By demonstrating protective effects in a ciliopathy model, this study suggests that GLP-1 RAs may confer benefits beyond appetite suppression in patients with genetic obesity syndromes affecting ciliary function, further broadening the mechanistic rationale for targeting this pathway across diverse genetic etiologies of obesity.
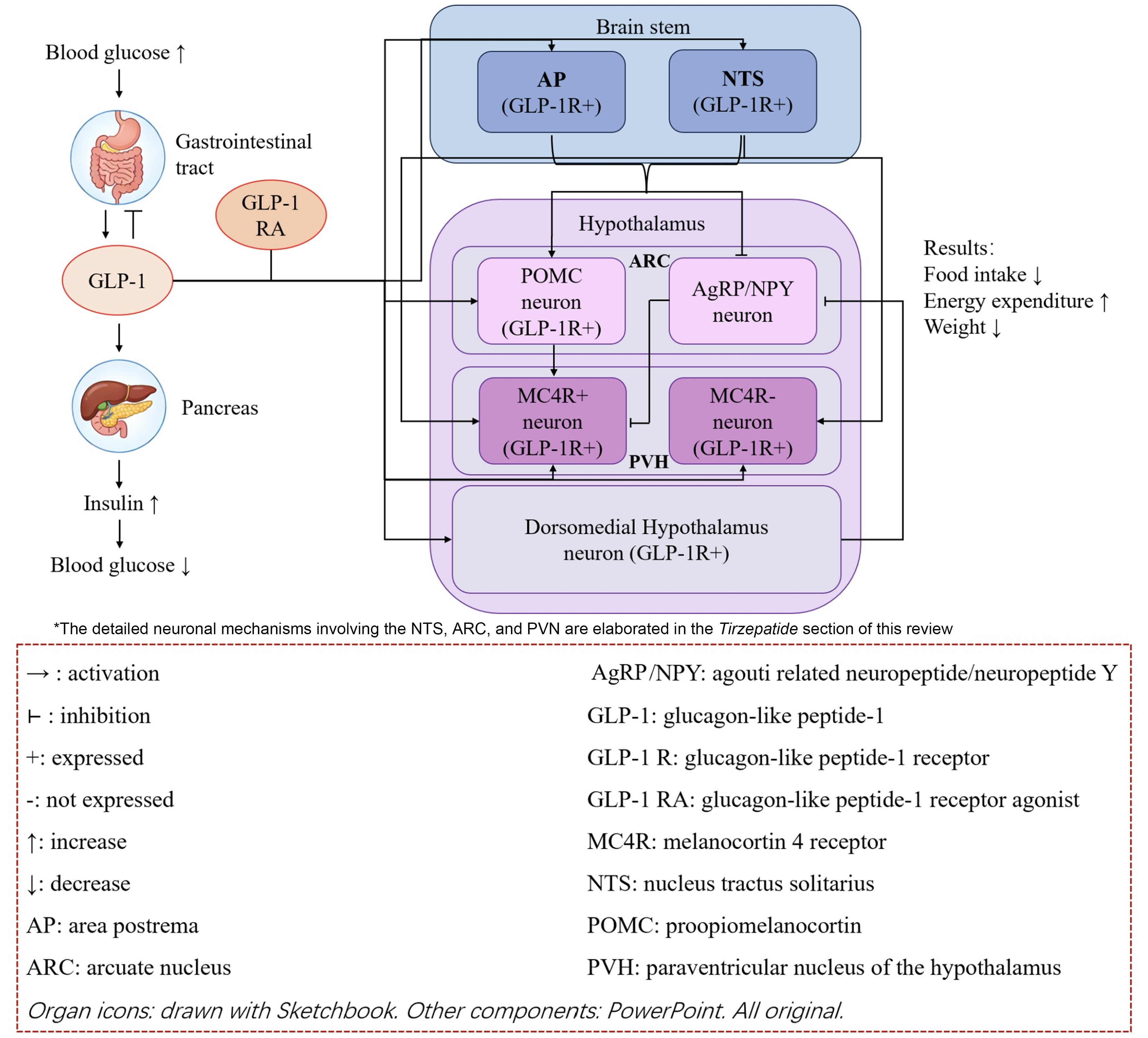
Figure 2. The GLP-1 axis and its relationship to the leptin-melanocortin pathway.
Liraglutide
Liraglutide is a long-acting GLP-1 RA approved both domestically and internationally for treating type 2 diabetes and for long-term weight management in obese or overweight patients aged 12 years and older. It is currently the most commonly used GLP-1 RA in research investigating the treatment of MC4R-deficient obesity. Adverse reactions include hypoglycemia and delayed gastric emptying, and it is not recommended for patients with medullary thyroid carcinoma or pancreatitis.
In 2018, Iepsen et al. first published clinical evidence on the efficacy of liraglutide in the treatment of MC4R-deficient obesity[67]. In their study, four severely obese patients diagnosed with pathogenic heterozygous MC4R variants (2 adults and 2 adolescents) achieved a mean weight reduction of 5.9 kg following 16 weeks of liraglutide treatment, corresponding to a 5.8% reduction from their baseline body weight. This observed effect appeared comparable to that observed in a control group of obese patients without MC4R variants, with the weight loss predominantly attributed to fat mass and minimal impact noted on lean body mass. Additionally, in a standardized meal test, MC4R variant carriers were reported to exhibit suppressed hunger, enhanced satiety, and a 28% reduction in energy intake during an ad libitum feeding test, with no significant differences compared to the control group. This study suggests that liraglutide may effectively reduce body weight and mitigate hyperphagia caused by MC4R deficiency, providing a basis for its potential therapeutic application in patients with MC4R-deficient obesity.
Regarding combination therapy, emerging evidence suggests that GLP-1 RAs may be an effective salvage treatment for post-surgical weight regain. Iepsen et al. reported a patient with a homozygous LOF MC4R variant (p.Arg165Gln) who initially lost 40 kg following Roux-en-Y gastric bypass but subsequently regained all lost weight[68]. After 16 weeks of liraglutide therapy, the patient achieved a 9.7 kg weight reduction, demonstrating that GLP-1 RAs can re-induce weight loss even after prior surgical failure[68]. Mechanistically, this complementarity is plausible: while surgery provides mechanical gastric restriction and alters gut hormone profiles, its inability to fully correct the underlying energy homeostasis dysregulation in MC4R deficiency often leads to long-term weight regain[43,45]. GLP-1 RAs, acting via MC4R-independent pathways in the brainstem and hypothalamus[62,63], may address this persistent hyperphagic drive that surgery alone cannot overcome. However, evidence-based algorithms guiding when and how to initiate GLP-1 RAs postoperatively are currently lacking. Whether initiating GLP-1 RAs earlier - either concomitantly with surgery or immediately upon detecting early weight regain - could improve long-term outcomes remains to be investigated.
For pediatric patients, a study investigating liraglutide treatment in a male with a homozygous LOF MC4R variant (c.750_751del p.Ile251fsTer34) - accompanied by severe insulin resistance, non-alcoholic fatty liver disease, and obstructive sleep apnea - has also indicated therapeutic benefits[69]. According to the report, despite the initiation of lifestyle modifications and metformin therapy at 9 years of age, the patient showed a poor response after 1 year of treatment: his BMI increased from 46.7 to 51.2 kg/m2, body fat percentage worsened from 53.1% to 65%, and he continued to experience difficulty achieving satiety with nocturnal awakening due to hunger and subsequent eating. Insulin resistance and liver enzyme levels further deteriorated. Given the circumstances, the patient was initiated on combination therapy with liraglutide and metformin. After 1 year of this treatment, a cessation of further gain was observed for the first time, with BMI decreasing to 50.3 kg/m2 - a level that remained elevated but suggested a halt in disease progression. Total body fat percentage slightly decreased (from 65% to 63.7%), and significant improvements were noted in insulin resistance, liver transaminase levels, and lipid profiles. Beyond appetite control, the patient also exhibited marked improvements in concentration and emotional stability, and participated more actively in extracurricular sports at school. This case report illustrates the potential role of liraglutide in weight management, appetite control, behavioral improvement, and metabolic regulation in pediatric patients with homozygous MC4R variants leading to complete pathway inactivation. It points to the versatility of liraglutide across different age groups, providing preliminary support for the treatment of early-onset MC4R-deficient obesity.
Regarding other therapeutic effects of liraglutide, a study found[70] that after 16 weeks of liraglutide treatment in 17 carriers of pathogenic heterozygous MC4R variants with a BMI > 28 kg/m2, both the variant group and control groups achieved significant and comparable weight loss (a 6% reduction from baseline). Additionally, liraglutide treatment appeared to significantly increase apparent bone mineral density in obese patients without MC4R variants in the control group, but no similar effect was observed in the MC4R-deficient group. No significant changes in bone turnover markers were noted in either group before or after treatment. These findings indicate that while liraglutide appears to preserve bone mass during weight loss, patients with MC4R-deficient obesity might represent an exception to this effect, though it does not seem to cause additional bone loss. This suggests that the bone-protective effect of liraglutide may require an intact MC4R signaling pathway. Thus, conceivably, additional bone-protective interventions may be necessary for obese patients carrying MC4R variants. Regarding other GLP-1 RAs, no published data currently exist on the bone effects of semaglutide or tirzepatide in MC4R deficiency. Preclinical and clinical studies in non-MC4R-deficient populations suggest that GLP-1 RA effects on bone metabolism may vary by agent, potentially due to differences in molecular structure, receptor affinity, or central vs. peripheral actions[71]. However, whether any GLP-1 RA can replicate the bone-sparing effects observed with liraglutide in common obesity - and whether such effects require intact MC4R signaling - remains unknown[6]. MC4R-deficient patients have distinct skeletal advantages at baseline; due to accelerated linear growth and increased mechanical loading from severe obesity, they typically exhibit higher bone mineral density compared to age- and BMI-matched individuals with common obesity[34]. This higher baseline density may provide a buffer against the bone loss invariably associated with significant weight reduction, potentially attenuating the clinical impact of losing MC4R-mediated bone protection during GLP-1 RA therapy. However, this buffering effect is unlikely to confer complete protection, and caution is warranted against assuming skeletal safety in the absence of monitoring. While no specific guidelines exist for MC4R-deficient patients, extrapolating from osteoporosis management principles suggests that baseline dual-energy X-ray absorptiometry (DXA) assessment may be reasonable in individuals with additional fracture risk factors or those anticipating substantial and sustained weight loss[72]. Periodic monitoring during therapy, particularly in patients who achieve ≥ 10% weight reduction, could help identify those who might benefit from bone-protective interventions. The potential role of adjunctive bone-protective therapies - including calcium and vitamin D supplementation, weight-bearing exercise, and, in selected high-risk cases, pharmacologic agents such as bisphosphonates - should be considered on an individual basis, though no evidence specifically addresses this question in MC4R-deficient patients. Ultimately, prospective studies evaluating bone outcomes with different GLP-1 RAs in this population, incorporating standardized DXA assessments and fracture surveillance, are needed to establish evidence-based skeletal health guidelines.
The therapeutic efficacy of liraglutide appears consistent regardless of the severity of the underlying MC4R deficiency. Both heterozygous patients with partial pathway impairment and homozygous patients with complete LOF mutations achieve clinically significant weight reduction (approximately 5%-10% from baseline) and experience marked reductions in hyperphagia. This robust effect across genotypes provides compelling evidence that liraglutide’s mechanism of action is largely independent of a functional MC4R pathway. Age-Related Considerations: The primary effect of appetite suppression and weight control is observed in both adults and pediatric patients. Nevertheless, the therapeutic goals and observed benefits may differ by age. In adults, the focus is predominantly on reversing weight regain and improving cardiometabolic parameters. In contrast, in pediatric cases with severe, life-threatening early-onset obesity, the primary achievement may be halting relentless disease progression and weight gain, a milestone in itself, alongside improvements in insulin sensitivity, liver enzymes, and notably, behavioral domains such as concentration and emotional stability. These findings underscore liraglutide’s versatility across genotypes and age groups, while also indicating that clinical expectations and adjunctive management strategies, particularly concerning bone health in MC4R-deficient patients, should be tailored accordingly.
Semaglutide
Semaglutide is a long-acting GLP-1 RA approved both globally to treat type 2 diabetes and long-term weight management in obese or overweight patients aged 12 years and older - consistent with liraglutide. Notably, its weight-reducing efficacy in patients with common obesity is superior to that of other GLP-1 RAs[73-75]. Additionally, it can reduce the risk of major adverse cardiovascular events in patients with pre-existing cardiovascular disease. Common adverse reactions include gastrointestinal symptoms (which may lead to intestinal obstruction in severe cases), headache, migraine, dehydration, and erectile dysfunction. Furthermore, semaglutide is not recommended for use in patients with acute pancreatitis or gallstones.
Preliminary evidence from the first published case report on semaglutide treatment for MC4R-deficient obesity[76] suggested a potential therapeutic effect. The report described a 13-year-old boy with a heterozygous LOF MC4R variant (p.Glu61Lys), accompanied by autism spectrum disorder and trypanophobia, who developed obesity at approximately 6 months of age. After the reported ineffectiveness of lifestyle modifications combined with metformin therapy initiated at age 9, semaglutide treatment was started at age 13. Following 1 year of therapy, significant reductions were observed in his body weight (from 187.5 to 166.9 kg), BMI (from 56.9 to 48.1 kg/m2), and body fat percentage (from 63.9% to 45.6%). Improvements were also noted in lipid profiles, blood glucose levels, and insulin resistance, along with substantial enhancements in quality of life and appetite control. Although transient elevations in liver metabolic enzymes occurred during treatment, subsequent abdominal ultrasound examinations revealed no hepatic abnormalities.
However, findings from another case report investigating semaglutide in MC4R-deficient obesity[77] indicated variability in treatment response. After 6 months of concurrent semaglutide treatment in an obese sibling pair carrying a heterozygous MC4R variant (c.508A>G, p.Ile170Val), only the sister exhibited modest improvements in weight-related parameters. While the brother experienced a reduction in BMI, his body weight and body fat percentage continued to increase, demonstrating different therapeutic responses compared to the previously reported cases. Notably, the report suggested that semaglutide might improve obesity-associated subclinical neurodegeneration and might promote the regeneration of small corneal nerve fibers, indicating a potential neuroprotective effect during treatment.
The potential causes of the differential therapeutic effects between the siblings may include genetic background and sex factors. First, although both siblings carry the same heterozygous MC4R variant, monogenic obesity is often modulated by polygenic interactions. A recent study found that variants in the GLP1R gene, such as rs6923761 G→A, can significantly affect the weight loss response to semaglutide[78]. It is plausible that the two siblings may carry different GLP1R genotypes: the sister might harbor the rs6923761 AA variant, which is associated with a higher weight loss rate, while the brother may carry the GG or GA variant that weakens semaglutide efficacy. In addition, the fat mass and obesity-associated gene (FTO), a well-recognized obesity-susceptibility gene, plays a crucial role in regulating semaglutide’s efficacy by modulating appetite and energy metabolism. A 5-year longitudinal study showed that FTO gene polymorphisms can lead to premature rebound of ghrelin (an appetite-promoting hormone) and a hypersensitive brain response to food stimuli, thereby weakening the appetite-suppressing effect of weight loss interventions[79]. In MC4R-deficient individuals, FTO risk allele carriers have significantly reduced weight loss efficacy after semaglutide treatment compared with those with normal genotypes, and the rate of weight regain in the later stage of treatment is significantly higher[79]. This suggests that if the brother carries FTO risk alleles while the sister does not, it may further blunt the brother’s response to semaglutide, resulting in continued increases in body weight and body fat percentage despite BMI reduction. Second, sex differences play an independent role in modulating semaglutide response. A prospective study involving 112 patients with severe obesity treated with semaglutide demonstrated that sex is a key predictor of therapeutic effect, with women showing significantly higher weight loss rates than men[78]. Specifically, women homozygous for the GLP1R rs6923761 A allele achieved a weight loss rate more than double that of men carrying the G allele[78]. More importantly, a large-scale 3b-phase randomized controlled trial (SURMOUNT-5), which compared semaglutide with tirzepatide in 751 obese adults without type 2 diabetes, explicitly reported that male patients had an average weight loss approximately 6 percentage points lower than female patients after 72 weeks of semaglutide treatment, further verifying that sex is an independent factor affecting semaglutide’s efficacy[80]. In the case, the sister’s modest weight improvement may benefit from female-specific hormonal environments (e.g., estrogen) that enhance GLP-1 receptor signaling, while the brother’s limited response could be related to male-specific factors that blunt semaglutide-induced appetite suppression and energy expenditure regulation.
Tirzepatide
Tirzepatide is currently the only long-acting glucose-dependent insulinotropic polypeptide (GIP)/GLP-1 dual receptor agonist. Its clinical evidence for weight loss demonstrates superior efficacy compared to GLP-1R mono-agonists, a difference rooted not merely in additive peripheral effects but in central neural synergy. This mechanistic advantage provides greater clinical confidence that it can achieve and sustain significant weight reduction. GIP receptors (GIPRs) are highly enriched in oligodendrocytes within the hypothalamic median eminence, and activation of these GIPRs regulates oligodendrocyte survival and myelin plasticity - this process enhances the entry of GLP-1R agonists into the brain via two key pathways: upregulating vascular endothelial growth factor A (VEGF-A) to increase blood-brain barrier permeability and facilitating GLP-1R agonist traversal of myelinated neuronal axons to target hypothalamic nuclei involved in energy homeostasis, including the ARC and PVN[81]. This GIP-mediated enhancement of central GLP-1 delivery is a unique feature not available to mono-agonists, ensuring a more potent engagement of central pathways. Upon entering the ARC, GLP-1 directly activates POMC neurons via its receptor. GIP signaling further upregulates the expression of the POMC gene in these neurons and amplifies their intracellular cAMP/protein kinase A (PKA) signaling pathway, thereby enhancing the production of satiety signals. Concurrently, GLP-1 and GIP act synergistically to potently and persistently suppress the AgRP neurons - an effect significantly more pronounced than with GLP-1 alone, leading directly to a reduction in hunger sensation and food intake. At this stage, the combined inhibitory effect of the dual signals on this neuronal population is significantly more potent than that achieved by GLP-1R mono-agonists. The POMC and AgRP neurons in the ARC, now under this coordinated regulation, project the integrated signals to downstream regions, such as the PVN[82]. Notably, studies in mice demonstrate that GLP-1R agonists specifically localize to and require arginine vasopressin neurons in the PVN to exert their hypophagic and weight-reducing effects, a process that is amplified by GIPR co-activation[81].
Rapid appetite suppression is also dependent on the coordinated regulatory functions of specific brainstem regions. The core process originates in the area postrema (AP), a circumventricular organ located outside the blood-brain barrier. The AP densely co-expresses the GIPR and the GLP-1R, enabling it to directly receive and integrate dual hormonal signals from the systemic circulation. The convergence of GIP and GLP-1 signaling at this site leads to synergistic amplification through an intracellular pathway, generating a potentiated initial satiety signal. This amplified signal is subsequently integrated within the nucleus tractus solitarius (NTS) with vagal afferent input from the gastrointestinal tract, forming a unified anorexigenic command. This command is then rapidly relayed to PVN via direct projections from the NTS. These findings provide a rapid neuroanatomical basis for the superior efficacy of GIP and GLP-1 dual receptor agonists[83].
In addition to this potent central appetite suppression, dual agonism also exerts complementary effects on energy expenditure[84]. The activation of central GIP signaling pathways has been linked to enhanced brown adipose tissue thermogenesis and the “browning” of white adipose tissue, promoting efficient fat mobilization[85]. Concurrently, preclinical studies have shown that the co-administration of GIP and GLP-1 into the cerebral ventricles enhances additional weight reduction benefits[86]. This multi-pathway approach - enhancing central access, synergizing at neuronal targets, and activating complementary energy expenditure pathways - provides a more robust physiological foundation than GLP-1 mono-agonism, justifying the stronger clinical evidence and the higher confidence in its substantial efficacy profile.
Tirzepatide has been approved for marketing in China and is indicated for long-term weight management in patients with obesity or overweight. Common adverse reactions include nausea, belching, acid reflux with heartburn, diarrhea, vomiting, constipation, abdominal discomfort, fatigue, injection site reactions, and hypersensitivity reactions. Since tirzepatide has been shown to induce medullary thyroid carcinoma in both in vivo rat models and in vitro studies, it is not recommended for use in patients with a history of medullary thyroid carcinoma or those with multiple endocrine neoplasia type 2 syndrome.
Results from a post hoc subgroup analysis of the SURMOUNT-1 study (NCT04184622), published this year[87], demonstrated that among 32 pathogenic MC4R variant carriers (1.4% of the study population) with a median age of 41.06 years and a BMI ≥ 40 kg/m2, treatment with tirzepatide for 72 weeks resulted in an 18.3% reduction in body weight from baseline. This weight loss was not statistically different from that observed in non-carriers (19.9% weight reduction). While these findings represent the most robust evidence to date for GLP-1 RA therapy in MC4R-deficient obesity, they derive from an exploratory analysis not pre-specified as a primary endpoint, and confirmation in prospective, dedicated trials is warranted. Additionally, tirzepatide appeared to yield similar improvements in metabolic parameters such as blood glucose, blood pressure, and lipid profiles among MC4R variant carriers and non-carriers, suggesting its efficacy in MC4R-deficient obesity may be comparable. Ongoing cohort studies (e.g., NCT06439277) are investigating the efficacy and safety of tirzepatide for weight management in adolescents, though further long-term studies are considered necessary to confirm these findings specifically in patients with MC4R-deficient obesity.
Notably, the current evidence base for GLP-1 RAs in MC4R-deficient obesity spans a wide age range, from children as young as 9 years to adults, yet pediatric-specific considerations remain inadequately addressed. Regulatory approvals differ by age: liraglutide and semaglutide are approved for adolescent obesity (≥ 12 years) in multiple jurisdictions, but their use in younger children represents off-label prescribing. Developmentally, severe hyperphagia profoundly impacts family dynamics and social functioning; while case reports suggest improvements in concentration and emotional stability following treatment, systematic assessment of quality-of-life outcomes remains lacking. The psychological impact of receiving a genetic diagnosis during childhood further necessitates integrated support alongside pharmacological intervention. Most critically, the paucity of pediatric-specific evidence cannot be overemphasized: the existing literature consists almost exclusively of case reports, with no completed RCTs specifically enrolling children with MC4R deficiency. Extrapolation from adult data cannot adequately address questions of optimal dosing, long-term safety across developmental stages, or effects on final adult height and peak bone mass, underscoring that dedicated pediatric research represents an urgent priority.
No direct comparative safety or tolerability data exist for GLP-1 RAs specifically in MC4R-deficient populations, nor are there systematic comparative studies in this group. The adverse effect profiles summarized above for liraglutide, semaglutide, and tirzepatide are derived entirely from studies in common obesity and type 2 diabetes. Whether these safety signals translate equivalently to MC4R-deficient patients - who are predominantly children and adolescents with a distinct metabolic phenotype - remains unknown. In the absence of population-specific data, clinical use should be accompanied by vigilant monitoring for class-wide effects (particularly gastrointestinal symptoms) and agent-specific concerns, with dose titration individualized to tolerability. Dedicated pharmacovigilance studies in this genetically defined cohort are urgently needed.
CONCLUSION
Therapeutic management of obesity due to MC4R deficiency represents a significant clinical challenge. Current mainstay interventions, including intensive lifestyle modification and bariatric surgery, demonstrate suboptimal and often unsustainable long-term efficacy in this genetically defined population, highlighting a critical unmet need for effective, accessible, and durable pharmacological strategies. This review synthesizes emerging evidence supporting the application of GLP-1 RAs as a viable and mechanistically rational alternative. Unlike MC4R-targeted agonists such as setmelanotide, whose efficacy is intrinsically limited by the functional status of the impaired receptor, GLP-1 RAs operate via pathways largely independent of the leptin-melanocortin circuitry. Preclinical and clinical data, though predominantly derived from case reports and small cohort studies to date, consistently indicate that GLP-1 RAs - including liraglutide, semaglutide, and the dual GIP/GLP-1 RA tirzepatide - can induce clinically meaningful weight loss, suppress the characteristic severe hyperphagia, and improve key cardiometabolic parameters in both heterozygous and homozygous MC4R variant carriers. Importantly, the weight-reducing efficacy appears comparable to that observed in common, polygenic obesity, as preliminarily evidenced by subgroup analyses from large trials such as SURMOUNT-1.
However, several key considerations must inform their clinical application. First, response heterogeneity exists, potentially influenced by factors such as the specific GLP-1 RA used, co-existing genetic modifiers (e.g., GLP1R or FTO variants), sex, and age. For instance, semaglutide may offer superior weight reduction in general obesity, but its response in MC4R deficiency requires further characterization. Tirzepatide, with its synergistic central mechanism, holds promise for potentially overcoming some of this heterogeneity and achieving greater efficacy. Second, the limited data suggest unique safety and monitoring aspects. The apparent absence of the bone mineral density preservation effect seen with liraglutide in MC4R-deficient patients, unlike in common obesity, underscores the need for vigilant bone health assessment and potential adjunctive interventions in this group. Third, the management goals may differ by age: in children and adolescents with early-onset, severe disease, halting relentless weight gain and improving metabolic and behavioral comorbidities may constitute primary success, whereas in adults, reversing weight regain and ameliorating cardiometabolic risk factors are central.
In conclusion, GLP-1-based therapy emerges not merely as a “promising” option but as a currently available, evidence-supported pharmacological strategy for MC4R-deficient obesity in regions where these agents are accessible. It addresses the core pathophysiological drivers - hyperphagia and impaired satiety - via a bypass mechanism. To translate this potential into optimized clinical practice, several research priorities should be emphasized. Prospective trials specifically enrolling MC4R-deficient patients are urgently needed, with stratification by mutation type - heterozygous vs. homozygous, partial vs. complete LOF - to delineate differential treatment responses. Given that this condition predominantly affects children, dedicated pediatric registries and long-term studies are essential to address the paucity of data on developmental safety, effects on linear growth and pubertal progression, and long-term cardiometabolic outcomes. The exploration of combination strategies, such as GLP-1 RAs with setmelanotide in patients retaining partial MC4R function, may offer additive benefits by targeting both MC4R-dependent and MC4R-independent pathways. Identification of predictive biomarkers, including GLP1R and FTO variants as noted above, could further guide personalized agent selection in this genetically heterogeneous population. Until targeted gene-specific therapies become widely available and effective for all MC4R variants, GLP-1 RAs represent a crucial therapeutic tool, offering a path toward significant clinical benefit for patients grappling with this challenging form of monogenic obesity.
The evidence reviewed herein does not yet support formal treatment guidelines, given the predominance of low-level data and the absence of head-to-head trials in MC4R-deficient populations. Nonetheless, several provisional considerations may assist clinical decision-making. Among currently available agents, GLP-1 RAs - and tirzepatide in particular, based on the SURMOUNT-1 subgroup analysis - have the most substantial weight-loss evidence in this population, with efficacy appearing comparable to that observed in common obesity. Liraglutide and semaglutide represent reasonable alternatives, supported by consistent signals from case-level data. Setmelanotide, while mechanistically rational, lacks confirmatory trial data specifically for MC4R-deficient obesity and should be regarded as an exploratory option at present. Critically, these considerations are offered as provisional clinical guidance only, not as formal recommendations, and must be individualized based on patient factors, drug availability, and regional regulatory status.
DECLARATIONS
Authors’ contributions
Performed literature search, review writing, data analysis, and interpretation: Ding M
Made significant contributions to the conception, design, and revision of the manuscript: Luo Y
Proposed the rationale and framework of review, provided guidance for manuscript design: Ji L
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool DeepSeek (version 3.2, released 2025-12-01) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work is funded by the 2024 National Clinical Key Specialty Construction Program of China (Department of Endocrinology, Peking University People’s Hospital) with support from the central government budget.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Mahmoud R, Kimonis V, Butler MG. Genetics of obesity in humans: a clinical review. Int J Mol Sci. 2022;23:11005.
2. Chami N, Wang Z, Svenstrup V, et al. Genetic subtyping of obesity reveals biological insights into the uncoupling of adiposity from its cardiometabolic comorbidities. Nat Med. 2025;31:3801-12.
3. Farooqi IS. Genetic and hereditary aspects of childhood obesity. Best Pract Res Clin Endocrinol Metab. 2005;19:359-74.
4. Singh RK, Kumar P, Mahalingam K. Molecular genetics of human obesity: a comprehensive review. C R Biol. 2017;340:87-108.
5. Duis J, Butler MG. Syndromic and nonsyndromic obesity: underlying genetic causes in humans. Adv Biol. 2022;6:e2101154.
6. Andermann ML, Lowell BB. Toward a wiring diagram understanding of appetite control. Neuron. 2017;95:757-78.
9. Balthasar N, Dalgaard LT, Lee CE, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493-505.
10. Wu Q, Palmiter RD. GABAergic signaling by AgRP neurons prevents anorexia via a melanocortin-independent mechanism. Eur J Pharmacol. 2011;660:21-7.
11. Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci. 2008;11:998-1000.
12. Huvenne H, Dubern B, Clément K, Poitou C. Rare genetic forms of obesity: clinical approach and current treatments in 2016. Obes Facts. 2016;9:158-73.
13. Mohammed I, Ahmed WS, Al-Barazenji T, Dauleh H, Love DR, Hussain K. Novel SIM1 variants expanding the spectrum of SIM1-related obesity. Int J Mol Sci. 2026;27:533.
14. Dietrich J, Lovell S, Veatch OJ, Butler MG. PHIP gene variants with protein modeling, interactions, and clinical phenotypes. Am J Med Genet A. 2022;188:579-89.
15. Li Y, Kim MH, Jiang L, et al. SH2B1 defends against energy imbalance, obesity, and metabolic disease via a paraventricular hypothalamus→dorsal raphe nucleus neurocircuit. Adv Sci. 2024;11:e2400437.
16. Polex-Wolf J, Yeo GS, O’Rahilly S. Impaired prohormone processing: a grand unified theory for features of Prader-Willi syndrome? J Clin Invest 2017;127:98-9.
17. Van Dijck E, Beckers S, Diels S, et al. Rare heterozygous PCSK1 variants in human obesity: the contribution of the p.Y181H variant and a literature review. Genes. 2022;13:1746.
18. Qian Y, Wu B, Liu R, et al. Case report: complete maternal uniparental isodisomy of chromosome 5 (iUPD(5)mat) with PCSK1 nonsense variant in an infant with recurrent diarrhea. Front Genet. 2021;12:668326.
19. Tao YX. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr Rev. 2010;31:506-43.
20. Loos RJF, Yeo GSH. The genetics of obesity: from discovery to biology. Nat Rev Genet. 2022;23:120-33.
21. Kleinendorst L, Massink MPG, Cooiman MI, et al. Genetic obesity: next-generation sequencing results of 1230 patients with obesity. J Med Genet. 2018;55:578-86.
22. Künzel R, Faust H, Bundalian L, et al. Detecting monogenic obesity: a systematic exome-wide workup of over 500 individuals. Int J Obes. 2025;49:1400-11.
24. Chami N, Preuss M, Walker RW, Moscati A, Loos RJF. The role of polygenic susceptibility to obesity among carriers of pathogenic mutations in MC4R in the UK Biobank population. PLoS Med. 2020;17:e1003196.
25. Hinney A, Schmidt A, Nottebom K, et al. Several mutations in the melanocortin-4 receptor gene including a nonsense and a frameshift mutation associated with dominantly inherited obesity in humans. J Clin Endocrinol Metab. 1999;84:1483-6.
26. Biebermann H, Krude H, Elsner A, Chubanov V, Gudermann T, Grüters A. Autosomal-dominant mode of inheritance of a melanocortin-4 receptor mutation in a patient with severe early-onset obesity is due to a dominant-negative effect caused by receptor dimerization. Diabetes. 2003;52:2984-8.
27. Stutzmann F, Tan K, Vatin V, et al. Prevalence of melanocortin-4 receptor deficiency in Europeans and their age-dependent penetrance in multigenerational pedigrees. Diabetes. 2008;57:2511-8.
28. Farooqi IS, Yeo GS, Keogh JM, et al. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest. 2000;106:271-9.
29. Hinney A, Volckmar AL, Knoll N. Melanocortin-4 receptor in energy homeostasis and obesity pathogenesis. Prog Mol Biol Transl Sci. 2013;114:147-91.
30. Zorn S, Bounds R, Williamson A, et al. Obesity due to MC4R deficiency is associated with reduced cholesterol, triglycerides and cardiovascular disease risk. Nat Med. 2025;31:4180-8.
31. Marx N, Husain M, Lehrke M, Verma S, Sattar N. GLP-1 receptor agonists for the reduction of atherosclerotic cardiovascular risk in patients with type 2 diabetes. Circulation. 2022;146:1882-94.
33. Kühnen P, Krude H, Biebermann H. Melanocortin-4 receptor signalling: importance for weight regulation and obesity treatment. Trends Mol Med. 2019;25:136-48.
34. Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085-95.
35. Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest. 2000;106:253-62.
36. Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD. The central melanocortin system can directly regulate serum insulin levels. Endocrinology. 2000;141:3072-9.
37. Hui H, Yu Y, Yiwei L, Li Y, Liling X, Dongguang Z. Genetic etiology and clinical features of non-syndromic pediatric obesity in the Chinese population: a large cohort study. BMC Pediatr. 2025;25:358.
38. Trier C, Hollensted M, Schnurr TM, et al. Obesity treatment effect in Danish children and adolescents carrying Melanocortin-4 Receptor mutations. Int J Obes. 2021;45:66-76.
39. Gong Y, Wu Q, Huang S, et al. Functional characterization of MC4R variants in Chinese morbid obese patients and weight loss after bariatric surgery. Adv Biol. 2023;7:e2300007.
40. Reinehr T, Hebebrand J, Friedel S, et al. Lifestyle intervention in obese children with variations in the melanocortin 4 receptor gene. Obesity. 2009;17:382-9.
41. Hainerová I, Larsen LH, Holst B, et al. Melanocortin 4 receptor mutations in obese Czech children: studies of prevalence, phenotype development, weight reduction response, and functional analysis. J Clin Endocrinol Metab. 2007;92:3689-96.
42. Zhu H, Yi X, He M, Wu S, Li M, Gao S. Exploring the interplay of genetic variants and environmental factors in childhood obesity: a systematic review and meta-analysis. Metabolism. 2025;170:156303.
43. Cooiman MI, Alsters SIM, Duquesnoy M, et al. Long-term weight outcome after bariatric surgery in patients with melanocortin-4 receptor gene variants: a case-control study of 105 patients. Obes Surg. 2022;32:837-44.
44. Fojas EGF, Radha SK, Ali T, Nadler EP, Lessan N. Weight and glycemic control outcomes of bariatric surgery and pharmacotherapy in patients with melanocortin-4 receptor deficiency. Front Endocrinol. 2021;12:792354.
45. Poitou C, Puder L, Dubern B, et al. Long-term outcomes of bariatric surgery in patients with bi-allelic mutations in the POMC, LEPR, and MC4R genes. Surg Obes Relat Dis. 2021;17:1449-56.
46. Campos A, Cifuentes L, Hashem A, et al. Effects of heterozygous variants in the leptin-melanocortin pathway on Roux-en-Y gastric bypass outcomes: a 15-year case-control study. Obes Surg. 2022;32:2632-40.
47. Zhang N, Wang H, Ran S, et al. Mutations in the leptin-melanocortin pathway and weight loss after bariatric surgery: a systematic review and meta-analysis. Obesity. 2024;32:1047-58.
48. U. S. Food & Drug Administration. FDA approves first treatment for weight management for people with certain rare genetic conditions. Available from: https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-first-treatment-weight-management-people-certain-rare-genetic-conditions. [Last accessed on 8 Jul 2026].
49. European Medicines Agency. Imcivree (setmelanotide): EPAR - product information. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/imcivree. [Last accessed on 8 Jul 2026].
50. Rodríguez Rondón AV, Welling MS, van den Akker ELT, et al. MC4R variants modulate α-MSH and setmelanotide induced cellular signaling at multiple levels. J Clin Endocrinol Metab. 2024;109:2452-66.
51. Phillips SA, van Santen HM, Hamilton JK, et al. OR11-01 efficacy and safety of setmelanotide in acquired hypothalamic obesity: results from a double-blind, multicenter, placebo-controlled, randomized phase 3 trial. J Endocr Soc. 2025;9:bvaf149.074.
52. Clément K, van den Akker E, Argente J, et al. ; Setmelanotide POMC and LEPR Phase 3 Trial Investigators. Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: single-arm, open-label, multicentre, phase 3 trials. Lancet Diabetes Endocrinol. 2020;8:960-70.
54. Clément K, Mosbah H, Poitou C. Rare genetic forms of obesity: from gene to therapy. Physiol Behav. 2020;227:113134.
55. Collet TH, Dubern B, Mokrosinski J, et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol Metab. 2017;6:1321-9.
56. Kühnen P, Clément K, Wiegand S, et al. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N Engl J Med. 2016;375:240-6.
57. Finimize. Rhythm Pharma’s Rare-Obesity drug keeps growth rolling. Available from: https://finimize.com/content/rytm-asset-snapshot. [Last accessed on 8 Jul 2026].
58. European Clinical Trials Database (EudraCT). Setmelanotide (RM-493) phase 2 treatment trial in patients with rare genetic disorders of obesity. Results for EudraCT number: 2017-000387-14. Available from: https://www.clinicaltrialsregister.eu/ctr-search/trial/2017-000387-14/results. [Last accessed on 8 Jul 2026].
59. AInvest. Rhythm pharmaceuticals’ $150M offering: fueling rare disease innovation amid dilution risks. Available from: https://www.ainvest.com/news/rhythm-pharmaceuticals-150m-offering-fueling-rare-disease-innovation-dilution-risks-2507/. [Last accessed on 8 Jul 2026].
60. Nauck MA, Wollschläger D, Werner J, et al. Effects of subcutaneous glucagon-like peptide 1 (GLP-1 [7-36 amide]) in patients with NIDDM. Diabetologia. 1996;39:1546-53.
61. Kim KS, Park JS, Hwang E, et al. GLP-1 increases preingestive satiation via hypothalamic circuits in mice and humans. Science. 2024;385:438-46.
62. Secher A, Jelsing J, Baquero AF, et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Invest. 2014;124:4473-88.
63. Dong Y, Carty J, Goldstein N, He Z, Hwang E, Chau D. Time and metabolic state-dependent effects of GLP-1R agonists on NPY/AgRP and POMC neuronal activity in vivo. Mol Metab. 2021;54:101352.
64. Velji-Ibrahim J, Radadiya D, Devani K, et al. Efficacy and safety of glucagon-like peptide-1 receptor agonists for obesity management in adults with and without type 2 diabetes: a systematic review. J Obes. 2025;2025:3897161.
65. Xie Z, Zheng G, Liang Z, Li M, Deng W, Cao W. Seven glucagon-like peptide-1 receptor agonists and polyagonists for weight loss in patients with obesity or overweight: an updated systematic review and network meta-analysis of randomized controlled trials. Metabolism. 2024;161:156038.
66. Eckert P, Nöller M, Müller M, et al. Targeting GLP-1 signaling ameliorates cystogenesis in a zebrafish model of nephronophthisis. Int J Mol Sci. 2025;26:7366.
67. Iepsen EW, Zhang J, Thomsen HS, et al. Patients with obesity caused by melanocortin-4 receptor mutations can be treated with a glucagon-like peptide-1 receptor agonist. Cell Metab. 2018;28:23-32.e3.
68. Iepsen EW, Have CT, Veedfald S, et al. GLP-1 receptor agonist treatment in morbid obesity and type 2 diabetes due to pathogenic homozygous melanocortin-4 receptor mutation: a case report. Cell Rep Med. 2020;1:100006.
69. Zaitoon H, Lubetzky R, Amir AZ, et al. Glucagon-like peptide-1 analog therapy in rare genetic diseases: monogenic obesity, monogenic diabetes, and spinal muscular atrophy. Acta Diabetol. 2023;60:1099-108.
70. Iepsen EW, Zhang J, Hollensted M, et al. Adults with pathogenic MC4R mutations have increased final height and thereby increased bone mass. J Bone Miner Metab. 2020;38:117-25.
71. Li X, Li Y, Lei C. Effects of glucagon-like peptide-1 receptor agonists on bone metabolism in type 2 diabetes mellitus: a systematic review and meta-analysis. Int J Endocrinol. 2024;2024:1785321.
72. Cosman F, de Beur SJ, LeBoff MS, et al. ; National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Osteoporos Int. 2014;25:2359-81.
73. O’Neil PM, Birkenfeld AL, McGowan B, et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet. 2018;392:637-49.
74. Ahmann AJ, Capehorn M, Charpentier G, et al. Efficacy and safety of once-weekly semaglutide versus exenatide ER in subjects with type 2 diabetes (SUSTAIN 3): a 56-week, open-label, randomized clinical trial. Diabetes Care. 2018;41:258-66.
75. Pratley RE, Aroda VR, Lingvay I, et al. ; SUSTAIN 7 investigators. Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): a randomised, open-label, phase 3b trial. Lancet Diabetes Endocrinol. 2018;6:275-86.
76. Gokul PR, Apperley L, Parkinson J, et al. Semaglutide, a long-acting GLP-1 analogue, for the management of early-onset obesity due to MC4R defect: a case report. Horm Res Paediatr. 2025;98:148-55.
77. Gad H, Mohammed I, Dauleh H, et al. Case report: nerve fiber regeneration in children with melanocortin 4 receptor gene mutation related obesity treated with semaglutide. Front Endocrinol. 2024;15:1385463.
78. Phan A, Carette C, Narjoz C, et al. A GLP1R gene variant and sex influence the response to semaglutide treatment in patients with severe obesity. Obesity. 2025;33:1237-42.
79. Li G, Hu Y, Zhang W, et al. FTO variant is associated with changes in BMI, ghrelin, and brain function following bariatric surgery. JCI Insight. 2024;9:e175967.
80. Aronne LJ, Horn DB, le Roux CW, et al. ; SURMOUNT-5 Trial Investigators. Tirzepatide as compared with semaglutide for the treatment of obesity. N Engl J Med. 2025;393:26-36.
81. Hansford R, Buller S, Tsang AH, et al. Glucose-dependent insulinotropic polypeptide receptor signaling in oligodendrocytes increases the weight-loss action of GLP-1R agonism. Cell Metab. 2025;37:1820-34.e5.
82. Jiang Y, Zhu H, Gong F. Why does GLP-1 agonist combined with GIP and/or GCG agonist have greater weight loss effect than GLP-1 agonist alone in obese adults without type 2 diabetes? Diabetes Obes Metab 2025;27:1079-95.
83. James-Okoro PP, Lewis JE, Gribble FM, Reimann F. The role of GIPR in food intake control. Front Endocrinol. 2025;16:1532076.
84. Liu J, Jing C, Guo Y, et al. The central signaling pathways related to metabolism-regulating hormones of the gut-brain axis: a review. J Transl Med. 2025;23:648.
85. Elmendorf AJ, Yousefian M, Kim IM, Hardaway JA, Habegger K, Flak JN. IUPHAR review: from foe to friend: Repurposing glucagon to treat obesity and type 2 diabetes. Pharmacol Res. 2026;223:108077.
86. Liu QK. Mechanisms of action and therapeutic applications of GLP-1 and dual GIP/GLP-1 receptor agonists. Front Endocrinol. 2024;15:1431292.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.