


Adeno-associated viral vector gene therapy for canine hemophilia
 0
0 
Abstract
Hemophilia A (HA) and B (HB) are monogenic X-linked disorders caused by plasma coagulant factor VIII (FVIII) and IX (FIX) deficiency, respectively, and occur naturally in a variety of dog breeds. While several animal models of hemophilia exist, the canine model closely mimics the genotypic, phenotypic, and immunologic characteristics of the disorder in humans. Consequently, decades of research were conducted in the canine model of hemophilia, ultimately leading to licensed liver-directed adeno-associated viral (AAV) vector-mediated gene therapy for HA and HB in humans. The studies from both research colony and companion hemophilic dogs with AAV liver-directed gene therapy support the safety and efficacy of this platform in canines. Here, we provide practical considerations with respect to the administration and monitoring of liver-directed AAV gene therapy for hemophilia in veterinary clinical practice and review the data on efficacy and safety of AAV gene therapy in canine hemophilia.
Keywords
INTRODUCTION
Hemophilia is an inherited bleeding disorder that occurs in humans and naturally in many animal species, including dogs, cats, cattle, horses, and sheep. Hemophilia A (HA) and B (HB) are caused by plasma coagulant factor VIII (FVIII) and IX (FIX) deficiency, respectively. Hemophilia is an X-linked recessive trait affecting primarily males, although females can experience bleeding with low coagulation factor levels resulting from skewed X chromosome inactivation in heterozygous female carriers, or, rarely, homozygous inheritance[1]. While there are several animal models of hemophilia, the genotypic, phenotypic, and immunologic characteristics of the naturally occurring canine model most closely mimic the disorder in humans[2-6]. Decades of research using the canine model of hemophilia ultimately led to clinical trials evaluating the safety and efficacy of liver-directed adeno-associated virus (AAV) vector-mediated gene therapy for HA and HB in humans, and canine models contribute to ongoing gene therapy development in other monogenic disorders[7]. In 2022, the US Food and Drug Administration (FDA) approved etranacogene dezaparvovec-drlb (Hemgenix®) as the first one-time gene therapy for adults with HB[8], followed by approval of valoctocogene roxaparvovec-rvox (Roctavian®) in 2023 for HA. Given that AAV gene therapy has proven to be a safe and effective treatment for research dogs with hemophilia, this novel therapy was offered to a limited number of companion dogs with HA and HB as part of an investigational study, with a 94% reduction in bleeding rates and an improved quality of life (QOL)[9,10]. There is a continued and growing interest in making gene therapy available to more companion dogs with hemophilia that exhibit a severe phenotype.
HEMOPHILIA IN DOGS
Clinical presentation and epidemiology
The bleeding phenotypes for both dogs and humans with hemophilia correlate with the degree of plasma coagulant factor deficiency. Both HA and HB are characterized as severe when factor activity is < 1% of normal, moderate for factor activity between 1% and 5%, and mild for factor activity between 5%-40%[11]. Dogs and humans with severe hemophilia exhibit frequent spontaneous bleeds, most commonly hematoma formation (in muscle or subcutaneous tissue), hemarthrosis, and oral bleeding associated with tooth loss or eruption, though other mucosal surface bleeding (e.g., epistaxis, gastrointestinal bleeding, hematuria) and hemorrhage into body cavities (thoracic pleural space, mediastinum, peritoneum, retroperitoneum) and the central nervous system can occur. While bleeding phenotypes can be variable in moderate and mild disease, moderate hemophilia typically results in traumatic or post-surgical bleeding with rarer spontaneous bleeding while those with mild deficiency generally have bleeding induced by surgery or trauma.
HA is far more common than HB in both dogs and humans. The mean estimated prevalence of HA and HB in humans is reported globally as 17.1 and 3.8 per 100,000 males, respectively[12]. There are insufficient data to determine the prevalence of hemophilia in dogs. Between 2018 and 2025, the Comparative Coagulation Laboratory at Cornell University diagnosed an average of 97 dogs per year with HA (~8-10 cases per month) and 8 dogs per year with HB (personal communication with Dr. Marjory Brooks, Comparative Coagulation Laboratory, Cornell University). Given that dogs with severe hemophilia might experience fatal hemorrhage or be euthanized before a diagnosis is confirmed and diagnostic evaluation might not be pursued for dogs with mild hemophilia due to infrequent bleeding events, the prevalence of canine hemophilia is likely under-recognized. Indeed, a prior veterinary study of FVIII-deficient dogs identified severe HA in 16 of 39 (41%) animals, four of whom were euthanized either because of their hemophilia diagnosis or due to severe bleeding despite transfusions[13].
Diagnosis
For dogs that exhibit a bleeding tendency, such as repeated spontaneous hemarthrosis or hematoma formation or excessive bleeding after surgery (e.g., scrotal hemorrhage post-castration) or trauma, initial diagnostic evaluation typically includes measurement of a platelet count, prothrombin time (PT), and activated partial thromboplastin time (aPTT). Dogs with HA or HB have a prolonged aPTT and normal PT, indicating an intrinsic coagulopathy. In a male dog, the most likely explanation for prolonged aPTT only is hemophilia. Plasma coagulation factor analysis is necessary to identify the specific factor deficiency. In the US, the Comparative Coagulation Laboratory at Cornell University is the reference laboratory for coagulation factor analysis. The required sample is citrate plasma. For accurate results, pre-transfusion blood samples should be obtained via clean venipuncture directly into sodium citrate tubes, with a citrate:blood ratio of 1:9. Factor VIII and FIX coagulant activities are measured using pooled canine FVIII- and FIX-deficient substrate plasma, respectively, in a modified aPTT assay. Results are reported as percentage FVIII and FIX coagulant activity compared to pooled normal canine plasma, which has an assigned value of 100% (reference interval, 50%-150%).
Although measurement of plasma FVIII and FIX coagulant activity is accurate for the diagnosis of hemophilia in dogs with a bleeding tendency, it does not always distinguish between carrier and clear females[14]. DNA testing can be performed for breeds with known disease-causing mutations to guide breeders with their mating decisions in an effort to eliminate the trait from their line[14-16]. The F8 and F9 genes are localized to the X chromosome in dogs and humans. The F8 gene is much larger than the F9 gene in both species, consisting of 26 and 8 exons, respectively, with the mature FVIII and FIX proteins having 2,332 (2,343 in the dog) and 415 (452 in the dog) amino acids, respectively. More than 4,000 F8 and 1,400 F9 variants have been identified in humans with HA and HB, respectively, and compiled in various global databases. In a genotyping study of more than 11,000 humans with hemophilia, F8 and F9 variants predicted to be gene-disrupting were common in severe hemophilia, while missense variants predominated in moderate and mild hemophilia[17]. Several disease-causing mutations have been identified in dogs with HA and HB [Table 1]. An intron 22 inversion of the F8 gene is the most common F8 mutation in both humans and dogs with severe HA[10].
Genetic variant identified in dogs with hemophilia
| Gene | Variant type | Variant | Breeds affected | References |
| F8 | Intron 22 inversion | Intron 22 inversion | Research colonies: Irish Setter, Miniature Schnauzer Companion dogs: Staffordshire terrier, French Bulldog, Labradoodle, Cocker Spaniel, Shih Tzu, mixed breed | [10,18,19] |
| F8 | Missense | Thr62Met | Shepherd mix | [10] |
| F8 | Small deletion | Exon 14 10-bp deletion | Mixed breed | [10] |
| F8 | Complex* | Complex* | Golden Retriever | [10] |
| F8 | Small deletion | Exon 14 2-bp deletion | Labrador Retriever | [20] |
| F8 | Nonsense | Exon 1 point mutation (TGG>TAG), Trp33Stop | German Shepherd | [21] |
| F8 | Nonsense | Exon 12 point mutation (CGA>TGA), Arg577Stop | Old English Sheepdog | [22] |
| F8 | Missense | Pro471Arg | Boxer | [16] |
| F8 | Missense | Cys548Tyr | German Shepherd | [16] |
| F8 | Small deletion | Exon 14 1-bp deletion p.Asn1069IlefsTer7 | Border Collie | [15] |
| F8 | SINE insertion | Exon 14 SINE insertion | Rhodesian Ridgeback | [23] |
| F8 | SINE insertion | Exon 14 SINE insertion (220 bp) | Havanese | [21] |
| F9 | Missense | Glu379Gly | Research colony: Cairn Terrier | [24] |
| F9 | Small deletion/transition | 5-bp deletion and transition p.Arg183Leufs*3 | Research colony: Lhasa Apso | [25] |
| F9 | Large deletion | Deletion of the entire gene | Labrador Retriever | [26] |
| F9 | Large deletion | Deletion 5’ to exon 6 | American Pit Bull Terrier | [27] |
| F9 | Insertion | Exon 8 5-kb insertion | Airedale Terrier | [27] |
| F9 | Insertion | Intron 5 1.5-kb insertion | German Wirehaired Pointer | [14] |
| F9 | Missense | Gly244Glu | Rhodesian Ridgeback | [28] |
| F9 | Small deletion | Single nucleotide deletion in the promoter | Hovawart | [29] |
| F9 | Insertion | Exon 8 insertion p.Asn274LysfsTer23 | Newfoundland | [30] |
| F9 | Missense | Gly12Arg; Ala28Thr | Boston terrier | [10] |
DNA testing for canine hemophilia is available for specific breeds through the Veterinary Genetics Laboratory at the University of California-Davis and the Comparative Coagulation Laboratory at Cornell University, as well as commercial dog DNA testing companies (e.g., Embark, Orivet, Wisdom Panel). Given that hemophilia is an X-linked recessive trait, there are 2 possible genotypes for male dogs: clear (normal F8 or F9 gene on X chromosome) or affected (F8 or F9 variant causing hemophilia on X chromosome). For females, there are 3 possible genotypes: clear (normal F8 or F9 gene on both X chromosomes), carrier (normal F8 or F9 gene on one X chromosome and F8 or F9 variant on the second X chromosome), or affected (F8 or F9 variant on both X chromosomes). Affected male and female dogs are typically evident based on their bleeding phenotype and plasma coagulant factor activity, but DNA testing is necessary for accurate detection of carrier females. Breeding a carrier female to a clear male will result in approximately 50% of the female offspring being carriers and 50% of the male offspring being affected with hemophilia. Therefore, carrier females should not be used for breeding.
Current standard of care in veterinary practice
Factor replacement therapy
The current standard of care for humans with severe HA and HB is prophylactic administration of recombinant human FVIII and FIX concentrates, respectively[31]. Patient compliance, poor venous access, and development of inhibitors are potential issues with prophylactic treatment with factor concentrates. Recombinant human FVIII and FIX concentrates have been administered to research dogs with HA and HB, respectively, and were efficacious in improving the results of hemostatic tests, but treated dogs developed inhibitory anti-human FVIII or FIX antibodies within 14 days[32,33]. While recombinant canine FVIII and FIX concentrates have been available in a research setting[10,34], there are no commercially available canine factor concentrates for use in clinical veterinary practice. Therefore, blood component therapy is the mainstay of managing severe bleeding. Furthermore, primarily due to expense, dogs with hemophilia are treated at the time of bleeding events rather than as a weekly prophylaxis. For dogs undergoing surgery, prophylactic protein replacement therapy is recommended immediately prior to surgery and then ideally every 8-12 h postoperatively for 1-2 days to provide adequate hemostasis. In general, it is more efficient to prevent bleeding than to react to bleeding postoperatively.
Canine blood products that contain functional coagulation factors include fresh whole blood (FWB), fresh-frozen plasma (FFP), cryoprecipitate (CRYO), and cryosupernatant or cryo-poor plasma (CPP). To maximize the use of each unit of FWB and minimize the risk of some transfusion-associated adverse events, ideally, the blood product selected should treat the specific need of the patient, i.e., replacement of plasma FVIII or FIX for HA or HB, respectively. FFP contains all coagulation factors and can be used to manage bleeding in dogs with HA or HB; therefore, it would be an appropriate choice for a male dog with a prolonged aPTT and presumptive diagnosis of hemophilia while awaiting results of coagulation factor analysis to determine if the dog has HA or HB. CRYO is prepared from FFP and is a concentrated product containing FVIII, von Willebrand factor (VWF), fibrinogen, fibronectin, and FXIII; it is the blood component of choice for management of bleeding in dogs with HA[35]. CPP contains FIX and other coagulation factors that are not present in CRYO, making it an excellent choice for management of bleeding in dogs with HB. If CRYO or CPP is not available, FFP is an appropriate choice for dogs with HA or HB. In addition, a red blood cell (RBC) transfusion (FWB or packed RBCs) might be necessary if an affected dog becomes moderately to severely anemic due to extensive bleeding.
The most serious complication of exogenous coagulation FVIII or FIX replacement is the development of inhibitors [immunoglobulin G (IgG) alloantibodies] that neutralize the function of infused factor replacement therapies. In both humans and dogs with hemophilia, the presence of an inhibitor is suspected when previously treated patients subsequently fail to respond clinically to protein replacement therapy. Inhibitors are measured by the Bethesda assay, with a positive inhibitor defined as a Bethesda titer of > 0.6 Bethesda units (BU)[11]. In humans, a low-responding inhibitor (defined as < 5.0 BU) tends to be transient and drop below threshold within 6 months, whereas a high-responding inhibitor (≥ 5.0 BU) tends to persist but may become undetectable after a long period without protein replacement therapy; however, inhibitor titers will increase 3-5 days after rechallenge with protein replacement[11].
In humans, inhibitors are more common in HA than HB and in severe hemophilia compared to mild to moderate disease. The reported incidence of inhibitors in humans with severe HA and HB is 30% and 12%, respectively, and with mild/moderate HA and HB 9% and 2%, respectively[17]. Other risk factors for the development of inhibitors in humans include high-impact (i.e., gene-disrupting) F8 or F9 variants, the type and intensity of protein replacement therapy, and, for HA, race/ethnicity[36-38]. Most of the information available regarding inhibitors in dogs is based on data from the HA dog colonies at the University of North Carolina at Chapel Hill (UNC-CH) and Queens University (QU) and the HB dog colonies at the University of Alabama (UAB) and UNC-CH[18,19,24,25]. Dogs with HA at both UNC-CH and QU have the intron 22 inversion F8 variant, yet the QU dogs, all from a common female ancestor, are considered “inhibitor-prone”, with approximately 25% developing inhibitors after protein replacement therapy. The UNC-CH dogs generally have a lower rate of inhibitor formation, although a separately bred subset of the colony is inhibitor-prone, indicating that genetic factors other than the disease-causing mutation impact the risk of inhibitor development[18,39,40]. The HB dogs at UAB have a F9 deletion variant and are “inhibitor-prone”, whereas the UNC-CH HB dogs have a F9 missense mutation and tend not to develop FIX inhibitors[39].
The incidence of inhibitors among companion dogs is largely unknown. Twelve dogs, 11 HA and 1 HB, were screened for inhibitors before adeno-associated viral (AAV) type 8 (AAV8) gene therapy[10]. Seven of 11 dogs had the intron 22 inversion mutation, and only 1 of these 7 dogs had a FVIII inhibitor; all other dogs tested negative[10]. Among other HA and HB dogs that have been genotyped, inhibitor screening was reported for only 2 dogs. An Old English Sheepdog with HA due to a nonsense mutation tested negative for FVIII inhibitors despite extensive treatment with FVIII to manage 55 bleeding events[22]. However, a Labrador Retriever with HB due to deletion of the entire FIX gene developed a low-responding inhibitor
Alternatives to factor replacement therapy
In clinical veterinary practice there are currently no alternatives to blood component therapy for management of bleeds in dogs with hemophilia. For human patients experiencing breakthrough bleeds despite prophylaxis, factor concentrates are recommended for those with no or low-titer inhibitors, while bypass agent therapy, either recombinant human FVIIa (rhFVIIa) or activated prothrombin complex concentrate (aPCC), is recommended for those with high-titer inhibitors[31]. Administration of rhFVIIa improved results of hemostatic tests in research dogs with HA (both with and without FVIII inhibitors) and HB, but the dogs developed anti-human FVIIa antibodies within approximately 2 weeks[41,42]. In a single research dog with HA, administration of recombinant canine FVIIa (rcFVIIa) was well tolerated and corrected the hemophilic coagulopathy[43]. Unfortunately, rcFVIIa is not commercially available. Therefore, for companion dogs that have developed FVIII or FIX inhibitors, supraphysiologic amounts of coagulation factor must be administered to overcome the inhibitors and provide hemostasis, with the negative consequence of further inhibitor formation.
Antifibrinolytics
Bleeding in hemophilia results from impaired clot formation, as well as premature lysis of the clot resulting from reduced clot stability due to the lack of amplified and persistent thrombin generation[44]. Administration of tranexamic acid (TXA), an antifibrinolytic that competitively inhibits the conversion of plasminogen to plasmin, combined with rhFVIII, increased clot resistance to accelerated fibrinolysis in humans with severe HA compared with factor replacement alone[45]. Similarly, in research dogs with HA, clot stability measured using thromboelastography in plasma samples collected at baseline and 4 h after intravenous administration of TXA (10 mg/kg) demonstrated delayed clot lysis at 4 h[46]. For humans with hemophilia, the World Federation of Hemophilia recommends antifibrinolytics as an alternative to use alone or as adjuvant treatment, particularly in controlling superficial soft tissue and mucosal bleeds (e.g., epistaxis, oral and gastrointestinal bleeding, and menorrhagia) and for dental surgery and eruption or shedding of teeth[31]. Similarly, minor bleeding events in dogs with hemophilia were controlled with TXA administered at 15-25 mg/kg orally every 6-8 h for approximately 5-7 days. Aminocaproic acid is an alternative antifibrinolytic, although it is used less widely in humans due to its shorter plasma half-life, lower potency, and higher toxicity compared to TXA[31]. For dogs with moderate to severe bleeding events that require protein replacement therapy, an antifibrinolytic can be administered as an adjunctive treatment.
Desmopressin
Desmopressin (1-deamino-8-D-arginine vasopressin, DDAVP) is a vasopressin type 2-receptor (V2R) agonist that causes endothelial cells to secrete stored VWF and FVIII into the bloodstream. Bleeding in humans with mild to moderate HA can be treated or prevented with DDAVP, which typically results in a 3-to 4-fold increase in basal FVIII levels in this patient population[47]. However, the response to DDAVP varies widely between individuals, with basal FVIII levels and the F8 gene defect influencing the response[47]. In contrast, the DDAVP-induced FVIII response in dogs is less robust, with normal dogs having a 1.3-1.5-fold increase in FVIII activity over basal levels[48,49]. There are limited data on efficacy of DDAVP in dogs with HA. Four dogs with mild HA (FVIII 10%-24%) treated with DDAVP (0.4-5 µg/kg SC) had a negligible increase in FVIII activity[50]. Not surprisingly, DDAVP administration (5 µg/kg IV) had no effect on FVIII activity in two research dogs with severe HA that had received retroviral vector gene therapy[49]. Desmopressin does not appear to be an effective adjunctive treatment in managing bleeding in dogs with HA, regardless of severity of disease.
Limitations compared to gene therapy
Factor replacement therapy, the mainstay of management of bleeding events in dogs with hemophilia, has a transient benefit. For dogs with severe HA or HB, some might require monthly transfusions to control spontaneous bleeds, resulting in a large financial burden for the client over time and, therefore, potentially euthanasia[13]. In contrast, AAV gene therapy for HA and HB provides a one-time treatment that yields a sustained increase in plasma FVIII or FIX activity for years (> 10 years documented in some research and companion dogs), a marked (94%) reduction in bleeding events, and an improved QOL[10].
LIVER-DIRECTED AAV GENE REPLACEMENT THERAPY IN CANINE HEMOPHILIA
As canine hemophilia closely mimics the human disease[2-6], studies in hemophilic dogs were informative in the development of HA/HB gene therapies for humans. Initial in vivo studies using retroviral or adenoviral vectors carrying either human or canine F8 and F9 complementary DNA (cDNA) were unsuccessful due to transgene silencing or the development of immune responses directed against either the vector, transduced cells, or the secreted xenoprotein that could not be rescued with immunosuppression[51-55]. Given these limitations, alternative vectors were investigated. Successful preclinical AAV vector gene therapy in other disease models[56,57] provided evidence to study AAV vectors in hemophilia. AAVs are single-stranded, nonpathogenic, replication-defective DNA viruses belonging to the Parvoviridae family[58]. The AAV genome is ~4.7 kb and comprises inverted terminal repeats (ITRs) surrounding replication and capsid genes; in vector manufacturing, these genes are replaced with the transgene and promoter of interest[59]. The capsid serotype provides some tissue specificity, which can be further refined by the promoter to limit transgene expression to the tissue of interest[60].
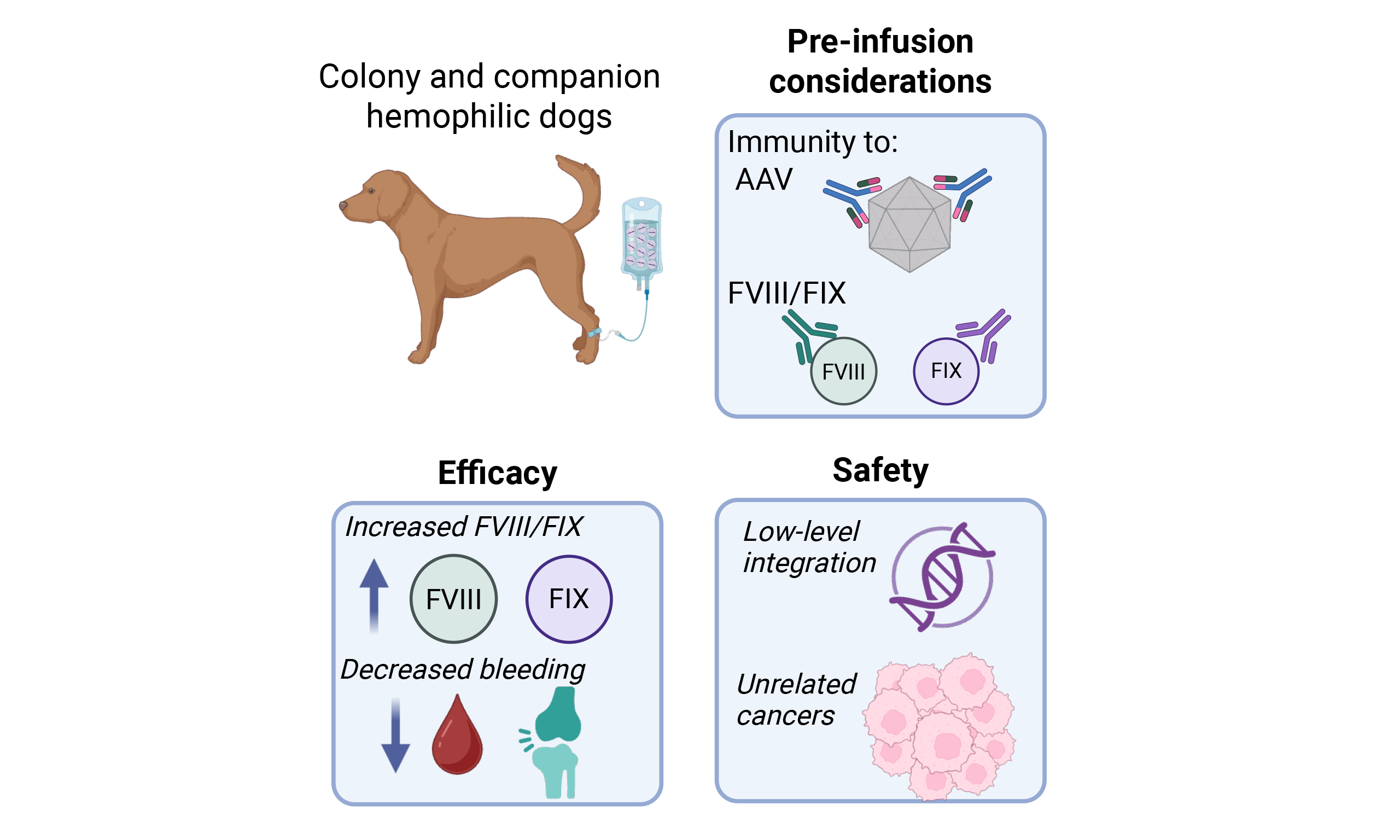
AAV vectors were initially studied in HB[61-63] owing to the smaller cDNA size of F9 (1.6 kb)[64] vs. full-length F8 (7 kb)[65,66] which could not be packaged into the AAV capsid. Identification of B-domain deleted (BDD) FVIII, which removed ~2.6 kb, in combination with other strategies, permitted investigation of these shortened F8 transgenes in AAV applications[67-70]. The ensuing decades of preclinical work[5,71,72] in animal models of hemophilia informed the development, and eventual licensure, of AAV vectors for HA and HB in people[8,73-77]. These preclinical canine studies were completed in dogs housed in research colonies[72,78-93]. A clinical study in companion dogs was also completed, informing on real-world safety and efficacy of AAV gene therapy in dogs with hemophilia[9,10]. Here, we provide practical considerations with respect to the administration and monitoring of liver-directed AAV gene therapy for hemophilia in veterinary clinical practice and review the data on efficacy and safety of AAV gene therapy in canine hemophilia that led to the clinical development and approval of AAV gene therapy for human hemophilia.
Pre-infusion and eligibility considerations
AAV-based gene therapy is largely indicated for dogs with severe disease (< 1% factor FVIII or FIX) to ameliorate the severe, recurrent bleeding events. While there is a mutational spectrum in canine hemophilia [Table 1], the outcomes after gene therapy do not appear to be impacted by the underlying genetic defect[9,10,18,19,22,72,82,90,93,94]. Therefore, functional activity assays to identify dogs with severe hemophilia have been sufficient to identify dogs that would be eligible to receive AAV vectors. However, proclivity for anti-FVIII or FIX inhibitors is associated with more severe genetic defects that result in no antigen production. Thus, testing for the presence of inhibitors prior to vector infusion for dogs exposed to replacement therapies and blood products is an important consideration.
Several studies in research colony-housed HA dogs have shown that liver-directed AAV gene therapy can both promote immune tolerance and provide sustained therapeutic FVIII levels. These studies have been completed in two primary canine HA research colonies (described above) with intron 22 inversion of F8[18,19]. The QU colony has historically been inhibitor-prone compared to the initially immune-tolerant colony at UNC-CH, though outbreeding of the UNC-CH colony has resulted in development of inhibitor-prone dogs there as well. More recently, the colony at UNC-CH has included a high responding inhibitor dog colony with a large gene deletion and emerging data from AAV gene therapy from this model suggests that AAV-mediated immune tolerance may be mutation and peak inhibitor titer-dependent[95]. Thus, the severity of the F8 mutation and the pre-AAV infusion anti-FVIII inhibitor titer may be important in predicting response to AAV gene therapy in the setting of an anti-FVIII immune response; this has also been borne in human disease[96-99]. In contrast to people with HB with FIX inhibitors, dogs with FIX inhibitors do not develop allergic symptoms or nephrotic syndrome[100]. Notably, HB dogs in research colonies with inhibitors have demonstrated the ability to induce tolerance to FIX, in addition to achieving therapeutic FIX levels, following AAV gene therapy with a hyperactive FIX variant (FIX-Padua)[72,93].
Another consideration for eligibility is the presence of anti-AAV antibodies at the time of infusion. Titers as low as 1:5 can ablate liver transduction following systemic delivery of AAV in mice[101], and a human patient with a 1:17 titer had a dramatic reduction in the efficacy of his gene therapy[102]. Yet, despite the high seroprevalence of anti-AAV antibodies in the human population[103], the same has not been seen in canines. The authors of this paper have worked with eight canine colonies covering four disease models and wild-type animals and have never had to exclude a dog for pre-existing anti-AAV antibodies. One of the large colonies in question at UNC-CH, run by Dr. Timothy Nichols, has only had one experiment impacted by anti-AAV antibodies (personal communication) despite decades of experience using AAV in dogs. In that instance, two dogs likely developed anti-AAV antibodies from exposure to treated animals shedding the vector (see section below on vector shedding)[104]. Notably, in our work to treat companion dogs with hemophilia with AAV, we have not had to exclude any animals. All twelve of the companion dogs we have treated had anti-AAV titers < 1:5 at the time of infusion[10]. Importantly, this is despite routine vaccination protocols in both the dog colony and companion animals that include the canine parvovirus vaccine.
Infusion and monitoring after AAV gene therapy
While portal vein injection was used in early canine studies, more recent studies deliver the AAV vectors via peripheral intravenous infusion, often using the cephalic or saphenous vein, in a sterile saline solution[10,94,105]. In both delivery methods, research colony and companion dogs received hemostatic therapy (either recombinant purified canine FVIII protein or FFP) immediately prior to AAV vector administration. Companion dogs were admitted and monitored in the hospital for 2 days following vector administration[9,10].
A short-term toxicity consideration in human AAV liver-directed gene therapy for hemophilia is the occurrence of a cellular immune response directed against the vector capsid. This capsid-directed response was not observed in preclinical murine and non-human primate AAV studies. However, it was noted in an early AAV type 2 (AAV2)-FIX gene therapy trial, in which approximately 4 weeks after vector infusion, a patient developed an asymptomatic rise in alanine aminotransferase (ALT) that coincided with a gradual loss of FIX expression over the ensuing 8-12 weeks[102]. Using an ELISPOT assay, that study showed evidence of activated T cells against the vector capsid, but not against FIX, confirming the hypothesis that transduced liver cells presenting the capsid were targeted for destruction by the host immune system. A follow-up study showed that corticosteroids could dampen the cellular immune response when a rise in ALT was observed and rescue FIX expression[73].
This capsid-directed cellular immune response has not borne out in canine hemophilia gene therapy studies with long-term follow-up[10,94,105]. In the UNC-CH colony dogs, ALT levels were measured two to four times per year and remained normal in 6/9 dogs at most of the timepoints analyzed. Two dogs who were older at time of vector infusion had minor ALT elevations (< 2 times upper limit of normal) starting 4 years after AAV administration and one dog had ALT elevations > 2 times the upper limit. However, there was no specific liver disease pattern (AST levels were within normal limits for all dogs) and serum α-fetoprotein (AFP) was normal at all timepoints. Postmortem examinations of the liver were conducted in both studies and compared to those of untreated age-matched dogs. In the QU research colony dogs, post-mortem examinations did not reveal any significant parenchymal inflammation, necrosis, hepatocyte injury, fibrosis, adenoma, or carcinoma[94]. In the UNC-CH colony dogs, there were no AAV-treated liver pathologic changes observed compared to untreated dogs[105]. In the companion dog study, there were no sustained elevations in AST or ALT across longitudinal follow-up and there was no evidence of decreased FVIII or FIX expression at times of transient elevations[10]. Together, these data suggest that hemophilic dogs do not experience the capsid-mediated cellular immune response noted in human subjects.
Unfortunately, no animal models faithfully recapitulate the anti-capsid cellular immune response seen in humans. While there are numerous hypotheses as to why this might be - including inbred strains having restricted major histocompatibility complex (MHC) haplotype pools[106], humans having previous exposures to wildtype AAVs[107], and humans having hyperactive T cells due to a CMP-N-acetylneuraminic acid hydroxylase (CMAH) mutation[108] - none have borne out. Outbred animal models including dogs and primates with broad MHC haplotypes do not mount such responses; previous vaccination against AAV still does not induce anti-capsid T cells in mice[107], and CMAH-/- mice do not have destruction of AAV-transduced hepatocytes[108].
Vector shedding guidance
In humans, AAV shedding in the urine is largely complete by one week after treatment[102]. Note that this “shedding” refers to detectable DNA sequences, not necessarily intact vector or vector that retains the ability to transduce. While it can persist longer in semen, in rabbits AAV shed in semen is no longer able to transduce by day 7[109], and in mice the shed vector may never be capable of transduction[110]. Nonetheless, in a recent study intending to treat HB dogs with intravenous AAV at 2 days, 30 days, 3 months, and 6 months of age, the animals treated at 30 days of life did not express detectable levels of FIX[104]. Interestingly, these dogs had frequent interactions with dogs treated at 2 days of life over the subsequent 28 days, a period during which the latter would be expected to shed vector (personal communication, Timothy Nichols, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA). Both dogs treated at 30 days were subsequently found to have anti-AAV antibodies. Due to the theoretical risks of vector shedding and the potential for acute adverse events, we have previously admitted companion animals for the 48 h after vector administration. However, given the lack of realized adverse events in human trials, the current plan is for future dogs to be treated in an outpatient setting, which may reduce costs associated with admission. Owners will be advised on hygiene care, likely with guidance similar to that given to the caregivers of young patients in diapers who receive Zolgensma®, an AAV gene therapy for spinal muscular atrophy. The suggested precautions for those caregivers are to wear gloves for diaper changes, double-bag the diaper, and clean hands thoroughly afterwards with soap and water or an alcohol-based hand sanitizer[111]. It is advised that diapers can still be disposed of in household waste.
Efficacy of liver-directed AAV gene replacement therapy in dogs
Hemophilia A
Thirty severe HA dogs without inhibitors have been reported in the literature [Table 2] with a median of 6.7 (range, 1.1-12) years of follow-up[9,10,83,88,92,94,105]. The dogs were treated with AAV2, AAV type 6 (AAV6), AAV8, and AAV type 9 (AAV9) vectors at doses ranging from 5.4 × 1012 to 5 × 1013 vg/kg. Efficacy measurements include the endogenous factor production level achieved as well as the reduction in bleeding rate normalized over a year (annualized bleeding rate or ABR). The goal of prophylaxis is to reduce spontaneous ABR to as close to zero as possible. The median canine FVIII activity by chromogenic assay was 3.4% (range, 0.3-13.8) and the median ABR was 0.1 (range, 0-2.3) bleeds per year. The median ABR before gene therapy in the QU colony was 4 (range, 2.1-10). Although the ABR was not reported in the UNC-CH colony study, those colony dogs typically experience 4-5 bleeds per year. In the companion dog study, the median ABR was 5.2 (range, 2.6-18.9) bleeds per year. The pre-treatment ABR trends higher in the companion dog study compared to the reported ABR in the QU colony study, suggesting that real-world activity may increase the risk of bleeding in the companion animal population. As HA is an X-linked disorder, most treated HA dogs were male. Across these reports, seven homozygous females were treated with AAV2, AAV6 and AAV8 vectors at doses ranging from 6 × 1012 to 3 × 1013 vg/kg. These females had a median FVIII activity of 2.25% (range, 0.3%-12.1%) and median ABR of 0.4 (range, 0-1.4) over a median of 8.2 (range, 1.1-11.5) years of follow-up, which is similar to their male counterparts at 3.5% FVIII activity and ABR of 0.2.
Efficacy of AAV canine gene therapy in non-inhibitor hemophilia A dogs
| Dog | Sex | Capsid | Transgene | Dose (vg/kg) | Follow-up (years) | FVIII activity (%) | ABR |
| Queens colony[83,94] | |||||||
| ELI | F | AAV2 | cFVIII-SQ | 6 × 1012 | 8.2 | 3.4 | 0.4 |
| VC | M | AAV2 | cFVIII-SQ | 1.5 × 1013 | 11 | 2.9 | 0.7 |
| JUN* | M | AAV2 | cFVIII-SQ | 2.7 × 1013 | 12 | 7.2 | 0.2 |
| ALX | F | AAV6 | cFVIII-SQ | 1 × 1013 | 10.5 | 8.6 | 0.0 |
| MZ | M | AAV6 | cFVIII-SQ | 1 × 1013 | 10.1 | 7.9 | 0.1 |
| FLO | F | AAV8 | cFVIII-SQ | 1 × 1013 | 10.5 | 1.8 | 0.1 |
| ANG | M | AAV2 | cFVIII-SQ | 1.5 × 1013 | 11.5 | 0.5 | 1.5 |
| MG | F | AAV6 | cFVIII-SQ | 1.7 × 1013 | 11.5 | 0.3 | 0.9 |
| UNC-CH colony[88,92,105] | |||||||
| F24 | M | AAV8 | Two chain | 2.5 × 1013 | 2.2 | 2.7 | 0.0 |
| Woodstock | M | AAV9 | Two chain | 2.5 × 1013 | 8.2 | 7.1 | 0.1 |
| J60 | M | AAV9 | Two chain | 2.5 × 1013 | 9.5 | 4.5 | 0.0 |
| Linus | M | AAV8 | Two chain | 1.2 × 1013 | 10.1 | 11.3 | 0.3 |
| H19 | M | AAV9 | Two chain | 1.2 × 1013 | 8.3 | 2.5 | 0.1 |
| M06 | M | AAV8 | cFVIII-SQ | 4 × 1013 | 2.3 | 9.4 | 0.4 |
| M50 | M | AAV8 | cFVIII-SQ | 4 × 1013 | 7.3 | 10.3 | 0.0 |
| L51 | M | AAV8 | cFVIII-SQ | 2 × 1013 | 6.1 | 1.9 | 0.2 |
| M66 | M | AAV8 | cFVIII-SQ | 2 × 1013 | 6 | 3.7 | 0.0 |
| G62 | F | AAV8 | Two chain | 1.52 × 1013 | 1.10 | 2-2.5 | 1-2 events |
| E73 | F | AAV8 | Two chain | 3 × 1013 | 1.78 | 2-2.5 | 1-2 events |
| Companion dogs[9,10] | |||||||
| PC1 | M | AAV8 | Two chain | 5 × 1013 | 8.88 | 2.4 | 1.7 |
| PC2 | M | AAV8 | Two chain | 5 × 1013 | 8.49 | 1.1 | 0.5 |
| PC3 | M | AAV8 | cFVIII-ΔF | 6 × 1012 | 2.56 | 2.1 | 2.3 |
| PC5 | M | AAV8 | cFVIII-ΔF | 6 × 1012 | 5.55 | 12.2 | 0.4 |
| PC6 | M | AAV8 | cFVIII-ΔF + V3 | 5.4 × 1012 | 5.03 | 13.8 | 0.0 |
| PC7 | M | AAV8 | cFVIII-ΔF + V3 | 6 × 1012 | 4.34 | 5.5 | 0.0 |
| PC8 | M | AAV8 | cFVIII-ΔF + V3 | 6 × 1012 | 3.41 | NA | 0.3 |
| PC9 | M | AAV8 | cFVIII-ΔF + V3 | 6 × 1012 | 3.29 | 3.2 | 0.0 |
| PC10 | F | AAV8 | cFVIII-ΔF + V3 | 9 × 1012 | 3.86 | 12.1 | 0.0 |
| PC11 | M | AAV8 | cFVIII-ΔF + V3 | 6 × 1012 | 3.77 | 1.5 | 1.3 |
| PC12 | M | AAV8 | cFVIII-ΔF + V3 | 6 × 1012 | 3.84 | 2 | 0.0 |
Hemophilia B
Initial studies for F9 gene delivery in HB dogs were conducted in skeletal muscle either via direct intramuscular injection[61,63,80] or transvenular delivery[85,91,112]. While these methods resulted in intramuscular expression, there was concern for immunogenicity with ectopic FIX expression. Liver-directed AAV gene therapy studies in canines were consequently done first with catheterization of the portal or mesenteric
Efficacy of AAV canine FIX gene therapy in non-inhibitor hemophilia B dogs
| Dog | Sex | Age (mo) | Capsid | Vector | Dose (vg/kg) | FIX antigen (ng/mL) | FIX activity (%) | Follow-up (years) | ABR |
| C51[86,113] | F | 6.5 | AAV2 | LSP-cFIX | 3.7 x 1011 | 3.6 ± 0.5 | NR | 1.0 | 0.0 |
| C52[86,113] | M | 6.5 | AAV2 | LSP-cFIX | 2.8 x 1011 | 2.9 ± 0.7 | 0.7 ± 0.2 | 3.2 | 0.9 |
| 45 | AAV5 | LSP-cFIX-W | 2.3 x 1013 | 799 | 16.0 ± 1.7 | 2.5 | 0 | ||
| C55[86,113] | M | 12 | AAV2 | LSP-cFIX-WPRE | 4.6 x 1012 | 218.1 ± 26.5 | 4 | 5.0 | 0.0 |
| D39[86,113] | F | 2.8 | AAV2 | LSP-cFIX-WPRE | 2.8 x 1012 | 31.7 ± 3.2 | < 0.1 | 2.7 | 2.2 |
| 36 | AAV8 | LSP-cFIX-W | 9.28 x 1012 | 785 | 15.7 ± 1.2 | 2 | 0 | ||
| Brad[82,89] | M | 9 | AAV2 | ApoE-hAAT-cFIX | 1.1 x 1012 | 473 | 10 | 7.5 | 0.0 |
| Semillon[82,89] | M | 5.5 | AAV2 | ApoE-hAAT-cFIX | 1.2 x 1012 | 167 | 5 | 8.0 | 0.0 |
| E34[82,89] | F | 5 | AAV2 | ApoE-hAAT-cFIX | 8 x 1011 | 262 ± 92 | 5 ± 2.5 | 1.0 | 0.0 |
| Beech[82,89] | M | 12 | AAV2 | ApoE-hAAT-cFIX | 3.4 x 1012 | < 2,560 | < 3 | 0.2 | Fatal |
| F56[84] | M | 3 | AAV2 | LSP-cFIX-WPRE | 2.2 x 1012 | < 5 | NR | 1.0 | 1.0 |
| G10[84] | M | 3 | AAV2 | LSP-cFIX-WPRE | 1.9 x 1013 | 30 ± 4.84 | NR | 0.6 | 0.0 |
| G43[86] | M | 7.6 | AAV8 | LSP-cFIX-W | 5.2 x 1012 | 1,300 ± 200 | 25.9 ± 4.7 | 1.3 | 0 |
| H12[86] | F | 4 | AAV8 | LSP-cFIX-W | 5.2 x 1012 | 468 ± 110 | 9.4 ± 2.2 | 1.2 | 0 |
| Wick[72] | M | AAV8 | ApoE-hAAT-cFIX-R338L (Padua) | 1 x 1012 | 3.33 ± 0.38 | 39.9 ± 5.8 | 1.6 | 0 | |
| Trex[72] | M | AAV8 | ApoE-hAAT-cFIX-R338L (Padua) | 3 x 1012 | 3.81 ± 1.29 | 26.8 ± 5.5 | 3 | 0 | |
| PC4[10] | M | 56 | AAV8 | cFIX-WT | 3 x 1012 | 7,255 | 8.7 | 6.04 | 0.0 |
Quality of life
The companion pet study did include a QOL assessment following gene therapy, albeit with a possibility of recall bias as the pre-gene therapy scoring was completed concurrently with post-gene therapy scoring[10]. Nevertheless, there was an increase in overall QOL including an improvement in nearly all negative attributes as well as positive physical attributes such as playfulness and activities of daily living[10]. Ongoing studies should include QOL assessments both before and at intervals after gene therapy to more formally capture improvements in companion dogs following gene therapy.
Comparison between research colony and companion dogs
Gene therapy has resulted in similar increases in factor activity and decreased bleeding rates in both research colony and companion dogs with hemophilia, despite differences in their disease-causing mutations and environment. The reported UNC-CH and QU colony dogs with severe HA had the intron 22 inversion mutation, as did 7 of 11 companion dogs. Four companion dogs (PC1, PC8, PC9, PC10) had F8 mutations that have not been previously described in dogs with hemophilia, similar to the heterogeneity of humans with severe HA. As noted above, the pre-treatment median ABR trended higher in the companion dogs (5.2) compared to the QU colony dogs (4). This is likely a reflection of their environment, with companion dogs having a more active lifestyle that increases the risk of bleeding events. The post-treatment median ABRs and FVIII activities were similar for both colony dogs (0.2% and 3.4%, respectively) and companion dogs (0.15% and 2.6%, respectively). Clinical monitoring is likely more intense for companion dogs, with owners closely observing dogs’ attitude, activity, and overall demeanor for any subtle changes that could be indicative of a bleeding or other adverse event. However, colony dogs are also closely monitored by dedicated staff who care for these dogs and are unlikely to be lost to follow-up. Whether a dog is living in a research colony or the real world, it is possible for bleeding events to go unnoticed. However, it is clear that gene therapy for severe hemophilia has resulted in a marked reduction in bleeding events in both colony and companion dogs. No notable differences in the safety of AAV gene therapy have been described between companion and research dog studies.
Safety of AAV gene replacement therapy in dogs
Immune responses to transgene, vector, and capsid
Anti-transgene immune responses have largely been transient in dogs without pre-existing immunity[83,87,92] with a notable exception in one of 4 HB dogs with a null F9 mutation treated with a liver-directed AAV vector[82]. For HA dogs, 1/9 in the QU colony[83,87] and 1/8 dogs in the UNC-CH colony[92] developed inhibitors at peak titers of 2.5-9 BU, which were eradicated by 7-9 weeks after vector injection. Notably, AAV liver-directed gene therapy has resulted in immune tolerance in dogs with pre-existing FVIII and FIX inhibitors [Table 4][10,72,90]. The possible mechanisms behind this tolerance have been discussed elsewhere[114], but likely include T regulatory cells[90,115,116]. These data largely support that liver-directed AAV gene therapy promotes tolerance to FVIII or FIX[117,118]; indeed, a clinical trial of AAV5-FVIII is ongoing in people with HA with inhibitors[119,120]. As discussed above, there have not been anti-capsid-directed cellular immune responses reported in hemophilic dogs treated with AAV. Of note, anti-AAV humoral immune responses against the injected capsid serotype have been robust and sustained at titers that would prohibit redosing[94,105].
Factor activity and inhibitor tolerance in hemophilic dogs
| Dog | Inhibitor titer (BU) | Time to inhibitor eradication (weeks) | Follow-up (years) | Factor activity (%) |
| Hemophilia A | ||||
| Wembley | 3.5 | 80 | 1.8 | < 1 |
| K01 | 12 | 5 | 1.9 | 1.5 |
| K03 | 12 | 4 | 2.4 | 8 |
| L44 | 4.5 | 4 | 1.3 | 1.5 |
| PC3 | 21 | 109 | 2.6 | 2.1 |
| Hemophilia B | ||||
| Wiley | 5 | 4 | 3.5 | 124.2 |
| Otis | 1.2 | 13 | 2.2 | 47.1 |
Serious adverse events
A few deaths were reported in the companion animal study, though none related to the vector itself. One dog (PC1) had neurologic deterioration at 9 years after gene therapy during an extensive trauma-induced forelimb hemorrhage at a FVIII activity of 2.4%. A second dog experienced a subdural hemorrhage 2 years after gene therapy with a FVIII activity of 2.1%. Recently, PC11 died following a presumed intracranial hemorrhage at 7 years following gene therapy; his last FVIII activity was 1.5% [Mary Beth Callan (MBC), unpublished data]. These deaths emphasize the need for higher steady-state factor levels to minimize the risk of serious bleeding events as all three dogs had levels < 3%. Clinical studies in moderate HA and HB mirror the potential for increased bleeding events, and recent guidelines in human disease suggest trough levels > 3%-5% to prevent bleeding complications[31]. This therapeutic level could be achieved using hyperactive transgenes which permit reduction of total AAV dose (thus limiting AAV-related toxicity) while enabling higher factor activity. Approaches to enhance FVIII activity are in various stages of development and evaluation[10,121]. For FIX, the FIX-Padua variant, with ~8-fold higher activity compared to wildtype protein[122], has demonstrated safety and efficacy in HB dogs[72] and humans[74,123,124], and it is the transgene of choice in most current AAV approaches.
Genotoxicity and insertional mutagenesis
Wildtype AAV can integrate into the host chromosome at a low rate into a specific site on chromosome 19 (AAVS1) in vitro[125,126]. Recent studies have reported an association of AAV2 with acute hepatitis in children[127,128], but there is no direct evidence for how AAV2 triggers hepatitis. However, recombinant AAV vectors only retain the ITRs from wildtype AAV and thus should have lower integration risk as they are largely maintained in episomal form in the host cell[60]. Integration of recombinant AAV has been described in human liver and, to a lesser degree, cardiac tissue[129], raising concern for tumorigenesis especially hepatocellular carcinoma. However, no causative role for wildtype[130-132] or recombinant[133,134] AAV integration in tumor development in humans has been clearly established.
Malignancies have been described both in human AAV trials[135-137] and in dogs[94,138]. One companion dog presented 3.2 years after gene therapy with fulminant multicentric lymphoma and was euthanized; integration analysis did not detect AAV in the tumor samples[138]. Examination for vector integration and oncogenic risk has been conducted in both the QU and UNC-CH colony HA dogs[94,105,139]. One dog in the QU colony developed a soft tissue tumor, which did not have evidence of detectable AAV copies, suggesting that it was unlikely related to vector administration[94]. Necropsy specimens from these dogs revealed no evidence of chronic liver disease or liver tumors[94]. Vector copies by quantitative polymerase chain reaction/reverse transcription polymerase chain reaction (qPCR/RT-PCR) were present in liver and spleen samples with canine FVIII messenger RNA only detected in the liver[94]. More sophisticated sequencing studies for AAV integration analysis from these specimens were subsequently conducted[139]. These specimens, obtained ~10 years from vector infusion, support that approximately 95% of the vector is retained in episomal form, with less than 5% that is integrated, with an integration frequency of 0.93 events per 1,000 cells. The majority of integration events were fragmented or rearranged and located in intergenic regions. Common integration sites were identified in proximity to 5 liver-expressed genes (KCNIP2, CLIC2, ABCB1, F8 and ALB) but no dysregulated expression of these genes was noted. No significant clonal expansion was seen in these samples and the distribution of integration sites proximal to transcriptional start sites of cancer genes was consistent with that of a random control.
In the UNC-CH colony, there were similarly no clinical adverse events or malignancy related to AAV administration detected in any of the 9 treated dogs[105]. Two dogs had gradual increase in FVIII expression starting at 4 years after AAV vector administration independent of transgene, vector dose, and promoter element thus raising the question of clonal expansion and vector integration. Neither dog had any inflammatory or hepatic pathologic findings. Sequencing analyses from 20 liver specimens in 6 dogs demonstrated 1,741 putative integration sites that were largely rearranged and fragmented (as in QU colony). Integration events were favored in transcriptional units near cytosine phosphate guanine islands and modestly more frequent in cancer-associated genes. Five genes were found at integration sites that expanded in multiple dogs: EGR2, EGR3, CCND1, LTO1, and ZNF365; with clusters noted at EGR2, EGR3, CCND1, ALB and DUSP1[105].
Notably, across both the QU and UNC-CH colonies, there were 5 overlapping integration site clusters noted (2 in chromosome 4 and 1 each in chromosomes 13, 18, and 25)[139]. Together, these studies suggest that integration of AAV vectors is a complex phenomenon with high variability and while clonal expansion may be seen in some dogs, there is no clear association with malignancy even after 10 years of follow-up. Ongoing investigations and long-term follow-up of dogs treated with AAV vectors will further inform on the genotoxicity of recombinant AAV vectors.
Limitations of current data
While the published data to date on the efficacy and safety of gene therapy for severe hemophilia in colony and companion dogs are laudable given the inherent expense and longevity of these studies, they comprise a relatively small sample size of a total of 52 dogs: 30 HA and 15 HB with no inhibitors, and 5 HA and 2 HB with inhibitors. Furthermore, while the colonies are outbred, the research colony dogs with hemophilia have the same disease-causing mutations. Given that the severity of the F8 mutation and the pre-AAV infusion anti-FVIII inhibitor titer may be important in predicting response to AAV gene therapy, inclusion of hemophilic dogs with different genetic backgrounds could be beneficial in future canine gene therapy studies.
FUTURE OF THE FIELD
With the commercialization of multiple AAV-based gene replacement therapies for HA and HB, preclinical research in canine models has slowed dramatically. The extremely sparse recently published research in dogs with hemophilia is focused on examining vector integration following AAV administration[105,138,139], long-term follow-up of animals treated years ago[10], or ways to modify gene expression after vector administration[140]. Unfortunately, commercially available AAV gene therapy products for human patients with hemophilia cannot be used in dogs due to the development of anti-human factor inhibitors in treated canines[52-54]. It is possible that the field will return to gene replacement studies in canines in order to solve the longevity issues seen in human patients. It is also possible that the next round of canine studies will focus on gene editing. As of yet, no studies have been published on using Clustered Regularly Interspaced Short Palindromic Repeats-based gene or base editing in dogs with hemophilia. Preclinical studies have been completed in mouse models of hemophilia[141-143] and in vitro work has shown that human liver cell lines with a mutated canine FIX gene knocked in can be corrected with gene editing[144]. However, even if AAV-based gene editing for hemophilia in research dogs and humans is developed, we expect that the ability to translate these products into widely available products for veterinary care will be similarly limited by the challenges faced by gene replacement therapies. The research-grade AAV vectors used in the canine hemophilia studies were produced by academic institutions or biotechnology companies. The vector manufacturing process is complex and time-consuming, with long turnaround times and at prohibitive costs for clinical-grade vectors (e.g., ~3 million US dollars (USD) wholesale acquisition cost for the commercial human vectors). FDA approval for gene therapy in dogs is managed by the FDA's Center for Veterinary Medicine (CVM) through the new animal drug application (NADA) process and would require veterinary studies for each vector/transgene.
CONCLUSIONS
Canine hemophilia has been an important large animal model for preclinical studies of gene therapy in hemophilia and other monogenic diseases. The major advantages over rodent models include the natural occurrence of hemophilia with genotypic heterogeneity in dogs of varying breeds, the presence of bleeding events similar to human disease, the recapitulation of inhibitor development following factor treatment, and the opportunity for long-term follow-up to inform on safety and efficacy. The studies described herein have informed the choice of vectors, transgenes, dosing, and safety in human trials, ultimately leading to the approval of AAV gene therapy vectors for HA and HB within the last 5 years. Whether and how AAV gene therapy can be brought to companion animals in clinical veterinary practice remains challenging. Overall, the cost of vector, manufacturing processes, and regulatory burden may prove too cumbersome to implement AAV gene therapy for hemophilia into clinical veterinary practice and thus limit gene therapy availability to research protocols. Nevertheless, the data shows that AAV gene therapy can be safely administered to companion animals with appropriate monitoring.
DECLARATIONS
Acknowledgment
The Graphical Abstract was created using BioRender (Doshi, B., 2026; available at https://BioRender.com/x1mpdqv).
Authors’ contributions
Conceptualized, wrote, and revised the manuscript: Doshi BS, Crudele JM, Callan MB
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
Not applicable.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Van Galen KPM, d'Oiron R, James P, et al. A new hemophilia carrier nomenclature to define hemophilia in women and girls: Communication from the SSC of the ISTH. J Thromb Haemost. 2021;19:1883-7.
2. Brinkhous KM, Sandberg H, Garris JB, et al. Purified human factor VIII procoagulant protein: comparative hemostatic response after infusions into hemophilic and von Willebrand disease dogs. Proc Natl Acad Sci USA. 1985;82:8752-6.
3. Giles AR, Tinlin S, Hoogendoorn H, et al. In vivo characterization of recombinant factor VIII in a canine model of hemophilia A (factor VIII deficiency). Blood. 1988;72:335-9.
4. Kingdon HS, Hassell TM. Hemophilic dog model for evaluating therapeutic effectiveness of plasma protein fractions. Blood. 1981;58:868-72.
5. Sabatino DE, Nichols TC, Merricks E, Bellinger DA, Herzog RW, Monahan PE. Animal models of hemophilia. Prog Mol Biol Transl Sci. 2012;105:151-209.
6. Graham JB, Buckwalter JA. Canine hemophilia; observations on the course, the clotting anomaly, and the effect of blood transfusions. J Exp Med. 1949;90:97-111.
7. Switonski M. Impact of gene therapy for canine monogenic diseases on the progress of preclinical studies. J Appl Genet. 2020;61:179-86.
8. Pipe SW, Leebeek FWG, Recht M, et al. Gene therapy with etranacogene dezaparvovec for Hemophilia B. N Engl J Med. 2023;388:706-18.
9. Callan MB, Haskins ME, Wang P, Zhou S, High KA, Arruda VR. Successful phenotype improvement following gene therapy for severe hemophilia A in privately owned dogs. PLoS ONE. 2016;11:e0151800.
10. Doshi BS, Samelson-Jones BJ, Nichols TC, et al. AAV gene therapy in companion dogs with severe hemophilia: Real-world long-term data on immunogenicity, efficacy, and quality of life. Mol Ther Methods Clin Dev. 2024;32:101205.
11. Blanchette VS, Key NS, Ljung LR, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12:1935-9.
12. Iorio A, Stonebraker JS, Chambost H, et al. Establishing the prevalence and prevalence at birth of hemophilia in males: a meta-analytic approach using national registries. Ann Intern Med. 2019;171:540-6.
13. Aslanian ME, Sharp CR, Rozanski EA, de Laforcade AM, Rishniw M, Brooks MB. Clinical outcome after diagnosis of hemophilia A in dogs. J Am Vet Med Assoc. 2014;245:677-83.
14. Brooks MB, Gu W, Barnas JL, Ray J, Ray K. A line 1 insertion in the Factor IX gene segregates with mild hemophilia B in dogs. Mamm Genome. 2003;14:788-95.
15. Brockmann M, Mensing N, von Luckner J, Müller E, Kehl A. Hemophilia A in a litter of Border Collies caused by a one base pair deletion in the F8 gene. Vet Clin Pathol. 2023;52:607-12.
16. Christopherson PW, Bacek LM, King KB, Boudreaux MK. Two novel missense mutations associated with hemophilia A in a family of Boxers, and a German Shepherd dog. Vet Clin Pathol. 2014;43:312-6.
17. Johnsen JM, Fletcher SN, Dove A, et al. Results of genetic analysis of 11 341 participants enrolled in the my life, our future hemophilia genotyping initiative in the United States. J Thromb Haemost. 2022;20:2022-34.
18. Hough C, Kamisue S, Cameron C, et al. Aberrant splicing and premature termination of transcription of the FVIII gene as a cause of severe canine hemophilia a: similarities with the intron 22 inversion mutation in human hemophilia. Thromb Haemostasis. 2017;87:659-65.
19. Lozier JN, Dutra A, Pak E, et al. The chapel hill hemophilia A dog colony exhibits a factor VIII gene inversion. Proc Natl Acad Sci USA. 2002;99:12991-6.
20. Hytönen MK, Viitanen S, Hundi S, Donner J, Lohi H, Kaukonen M. A frameshift deletion in F8 associated with hemophilia A in Labrador Retriever dogs. Anim Genet. 2023;54:606-12.
21. Mischke R, Wilhelm Ch, Czwalinna A, Varvenne M, Narten K, von Depka M. Canine haemophilia A caused by a mutation leading to a stop codon. Vet Rec. 2011;169:496b.
22. Lozier JN, Kloos MT, Merricks EP, et al. Severe hemophilia A in a male old english sheep dog with a C→T Transition that created a premature stop codon in factor VIII. Comp Med. 2016;66:405-11.
23. Kehl A, Haaland AH, Langbein-Detsch I, Mueller E. A SINE insertion in F8 gene leads to severe form of hemophilia A in a family of rhodesian ridgebacks. Genes. 2021;12:134.
24. Evans JP, Brinkhous KM, Brayer GD, Reisner HM, High KA. Canine hemophilia B resulting from a point mutation with unusual consequences. Proc Natl Acad Sci USA. 1989;86:10095-9.
25. Mauser A, Whitlark J, Whitney K, Lothrop CJ. A deletion mutation causes hemophilia B in Lhasa Apso dogs. Blood. 1996;88:3451-5.
26. Brooks MB, Gu W, Ray K. Complete deletion of factor IX gene and inhibition of factor IX activity in a Labrador Retriever with hemophilia B. J Am Vet Med Assoc. 1997;211:1418-21.
27. Gu W, Brooks M, Catalfamo J, Ray J, Ray K. Two distinct mutations cause severe hemophilia B in two unrelated canine pedigrees. Thromb Haemostasis. 2017;82:1270-5.
28. Mischke R, Kühnlein P, Kehl A, et al. G244E in the canine factor IX gene leads to severe haemophilia B in Rhodesian Ridgebacks. Vet J. 2011;187:113-8.
29. Brenig B, Steingräber L, Shan S, et al. Christmas disease in a Hovawart family resembling human hemophilia B Leyden is caused by a single nucleotide deletion in a highly conserved transcription factor binding site of the F9 gene promoter. Haematologica. 2019;104:2307-13.
30. Kuder H, Sandzhieva-Vuzzo L, Kehl A, Rappaport JM, Müller E, Giger U. A single base insertion in F9 causing hemophilia B in a family of newfoundland-parti standard poodle hybrid dogs. Genes. 2021;12:1491.
31. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26 Suppl 6:1-158.
32. Brinkhous K, Sigman J, Read M, et al. Recombinant human factor IX: replacement therapy, prophylaxis, and pharmacokinetics in canine hemophilia B. Blood. 1996;88:2603-10.
33. Littlewood JD, Barrowcliffe TW. The development and characterisation of antibodies to human factor VIII in haemophilic dogs. Thromb Haemostasis. 2018;57:314-21.
34. Sabatino DE, Freguia CF, Toso R, et al. Recombinant canine B-domain-deleted FVIII exhibits high specific activity and is safe in the canine hemophilia A model. Blood. 2009;114:4562-5.
35. Stokol T, Parry B. Efficacy of fresh-frozen plasma and cryoprecipitate in dogs with von Willebrand's disease or hemophilia A. J Vet Intern Med. 1998;12:84-92.
36. Pipe SW, Sabatino DE, Nugent DJ, et al. Executive summary of the NHLBI state of the science (SOS) workshop: overview and next steps in generating a national blueprint for future research on factor VIII inhibitors. Haemophilia. 2019;25:610-5.
37. DiMichele DM. Inhibitors in childhood hemophilia A: genetic and treatment-related risk factors for development and eradication. Pediatr Blood Cancer. 2013;60 Suppl 1:S30-3.
38. DiMichele D. Inhibitor development in haemophilia B: an orphan disease in need of attention. Br J Haematol. 2007;138:305-15.
39. Arruda VR, Samelson-Jones BJ. Gene therapy for immune tolerance induction in hemophilia with inhibitors. J Thromb Haemost. 2016;14:1121-34.
40. Nichols TC, Hough C, Agersø H, Ezban M, Lillicrap D. Canine models of inherited bleeding disorders in the development of coagulation assays, novel protein replacement and gene therapies. J Thromb Haemost. 2016;14:894-905.
41. Brinkhous KM, Hedner U, Garris JB, Diness V, Read MS. Effect of recombinant factor VIIa on the hemostatic defect in dogs with hemophilia A, hemophilia B, and von Willebrand disease. Proc Natl Acad Sci USA. 1989;86:1382-6.
42. Othman M, Powell S, Hopman WM, Lillicrap D. Variability of thromboelastographic responses following the administration of rFVIIa to haemophilia A dogs supports the individualization of therapy with a global test of haemostasis. Haemophilia. 2010;16:919-25.
43. Knudsen T, Kristensen AT, Nichols TC, et al. Pharmacokinetics, pharmacodynamics and safety of recombinant canine FVIIa in a study dosing one haemophilia A and one haemostatically normal dog. Haemophilia. 2011;17:962-70.
44. Broze GJ, Higuchi D. Coagulation-dependent inhibition of fibrinolysis: role of carboxypeptidase-U and the premature lysis of clots from hemophilic plasma. Blood. 1996;88:3815-23.
45. Hvas AM, Sørensen HT, Norengaard L, Christiansen K, Ingerslev J, Sørensen B. Tranexamic acid combined with recombinant factor VIII increases clot resistance to accelerated fibrinolysis in severe hemophilia A. J Thromb Haemost. 2007;5:2408-14.
46. Strilchuk AW, Hur WS, Batty P, et al. Lipid nanoparticles and siRNA targeting plasminogen provide lasting inhibition of fibrinolysis in mouse and dog models of hemophilia A. Sci Transl Med. 2024;16:eadh0027.
47. Guillet B, Pawlowski M, Boisseau P, et al. Genotype-dependent response to desmopressin in hemophilia A and proposal of a predictive response score. Thromb Haemostasis. 2024;124:922-36.
48. Sato I, Parry BW. Effect of desmopressin on plasma factor VIII and von Willebrand factor concentrations in Greyhounds. Aust Vet J. 1998;76:809-12.
49. Xu L, Nichols TC, Sarkar R, McCorquodale S, Bellinger DA, Ponder KP. Absence of a desmopressin response after therapeutic expression of factor VIII in hemophilia A dogs with liver-directed neonatal gene therapy. Proc Natl Acad Sci USA. 2005;102:6080-5.
50. Mansell PD, Parry BW. Changes in factor VIII: coagulant activity and von Willebrand factor antigen concentration after subcutaneous injection of desmopressin in dogs with mild hemophilia A. J Vet Intern Med. 1991;5:191-4.
51. Kay MA, Rothenberg S, Landen CN, et al. In vivo gene therapy of hemophilia B: sustained partial correction in factor IX-deficient dogs. Science. 1993;262:117-9.
52. Kay MA, Landen CN, Rothenberg SR, et al. In vivo hepatic gene therapy: complete albeit transient correction of factor IX deficiency in hemophilia B dogs. Proc Natl Acad Sci USA. 1994;91:2353-7.
53. Fang B, Eisensmith RC, Wang H, et al. Gene therapy for hemophilia B: host immunosuppression prolongs the therapeutic effect of adenovirus-mediated factor IX expression. Hum Gene Ther. 1995;6:1039-44.
54. Connelly S, Mount J, Mauser A, et al. Complete short-term correction of canine hemophilia A by in vivo gene therapy. Blood. 1996;88:3846-53.
56. Flotte TR, Afione SA, Conrad C, et al. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proc Natl Acad Sci USA. 1993;90:10613-7.
57. Kaplitt MG, Leone P, Samulski RJ, et al. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet. 1994;8:148-54.
58. Berns KI. The Gordon Wilson Lecture. From basic virology to human gene therapy. Trans Am Clin Climatol Assoc. 1999;110:75-85.
59. Doshi BS, Arruda VR. Gene therapy for hemophilia: what does the future hold? Ther Adv Hematol. 2018;9:273-93.
60. Wang JH, Gessler DJ, Zhan W, Gallagher TL, Gao G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduct Target Ther. 2024;9:78.
61. Herzog RW, Hagstrom JN, Kung SH, et al. Stable gene transfer and expression of human blood coagulation factor IX after intramuscular injection of recombinant adeno-associated virus. Proc Natl Acad Sci USA. 1997;94:5804-9.
62. Monahan PE, Samulski RJ, Tazelaar J, et al. Direct intramuscular injection with recombinant AAV vectors results in sustained expression in a dog model of hemophilia. Gene Ther. 1998;5:40-9.
63. Chao H, Samulski R, Bellinger D, Monahan P, Nichols T, Walsh C. Persistent expression of canine factor IX in hemophilia B canines. Gene Ther. 1999;6:1695-704.
64. Choo KH, Gould KG, Rees DJ, Brownlee GG. Molecular cloning of the gene for human anti-haemophilic factor IX. Nature. 1982;299:178-80.
65. Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature. 1984;312:326-30.
66. Wood WI, Capon DJ, Simonsen CC, et al. Expression of active human factor VIII from recombinant DNA clones. Nature. 1984;312:330-7.
67. Lind P, Larsson K, Spira J, et al. Novel forms of B-domain-deleted recombinant factor VIII molecules. Construction and biochemical characterization. Eur J Biochem. 1995;232:19-27.
68. McIntosh J, Lenting PJ, Rosales C, et al. Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood. 2013;121:3335-44.
69. Pittman DD, Alderman EM, Tomkinson KN, Wang JH, Giles AR, Kaufman RJ. Biochemical, immunological, and in vivo functional characterization of B-domain-deleted factor VIII. Blood. 1993;81:2925-35.
70. Siner JI, Iacobelli NP, Sabatino DE, et al. Minimal modification in the factor VIII B-domain sequence ameliorates the murine hemophilia A phenotype. Blood. 2013;121:4396-403.
71. High KA. Theodore E. Woodward Award. AAV-mediated gene transfer for hemophilia. Trans Am Clin Climatol Assoc. 2003;114:337-51; discussion 51.
72. Crudele JM, Finn JD, Siner JI, et al. AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood. 2015;125:1553-61.
73. Nathwani AC, Rosales C, McIntosh J, et al. Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol Ther. 2011;19:876-85.
74. George LA, Sullivan SK, Giermasz A, et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N Engl J Med. 2017;377:2215-27.
75. Rangarajan S, Walsh L, Lester W, et al. AAV5-factor VIII Gene transfer in severe hemophilia A. N Engl J Med. 2017;377:2519-30.
76. Pasi KJ, Rangarajan S, Mitchell N, et al. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N Engl J Med. 2020;382:29-40.
77. Ozelo MC, Mahlangu J, Pasi KJ, et al. Valoctocogene roxaparvovec gene therapy for hemophilia A. N Engl J Med. 2022;386:1013-25.
78. Snyder RO, Miao C, Meuse L, et al. Correction of hemophilia B in canine and murine models using recombinant adeno-associated viral vectors. Nat Med. 1999;5:64-70.
79. Arruda VR, Fields PA, Milner R, et al. Lack of germline transmission of vector sequences following systemic administration of recombinant AAV-2 vector in males. Mol Ther. 2001;4:586-92.
80. Herzog RW, Mount JD, Arruda VR, High KA, Lothrop CD Jr. Muscle-directed gene transfer and transient immune suppression result in sustained partial correction of canine hemophilia B caused by a null mutation. Mol Ther. 2001;4:192-200.
81. Herzog RW, Fields PA, Arruda VR, et al. Influence of vector dose on factor IX-specific T and B cell responses in muscle-directed gene therapy. Hum Gene Ther. 2002;13:1281-91.
82. Mount JD, Herzog RW, Tillson DM, et al. Sustained phenotypic correction of hemophilia B dogs with a factor IX null mutation by liver-directed gene therapy. Blood. 2002;99:2670-6.
83. Scallan CD, Lillicrap D, Jiang H, et al. Sustained phenotypic correction of canine hemophilia A using an adeno-associated viral vector. Blood. 2003;102:2031-7.
84. Harding TC, Koprivnikar KE, Tu GH, et al. Intravenous administration of an AAV-2 vector for the expression of factor IX in mice and a dog model of hemophilia B. Gene Ther. 2004;11:204-13.
85. Arruda VR, Stedman HH, Nichols TC, et al. Regional intravascular delivery of AAV-2-F.IX to skeletal muscle achieves long-term correction of hemophilia B in a large animal model. Blood. 2005;105:3458-64.
86. Wang L, Calcedo R, Nichols TC, et al. Sustained correction of disease in naive and AAV2-pretreated hemophilia B dogs: AAV2/8-mediated, liver-directed gene therapy. Blood. 2005;105:3079-86.
87. Jiang H, Lillicrap D, Patarroyo-White S, et al. Multiyear therapeutic benefit of AAV serotypes 2, 6, and 8 delivering factor VIII to hemophilia A mice and dogs. Blood. 2006;108:107-15.
88. Sarkar R, Mucci M, Addya S, et al. Long-term efficacy of adeno-associated virus serotypes 8 and 9 in hemophilia a dogs and mice. Hum Gene Ther. 2006;17:427-39.
89. Niemeyer GP, Herzog RW, Mount J, et al. Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood. 2009;113:797-806.
90. Finn JD, Ozelo MC, Sabatino DE, et al. Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood. 2010;116:5842-8.
91. Arruda VR, Stedman HH, Haurigot V, et al. Peripheral transvenular delivery of adeno-associated viral vectors to skeletal muscle as a novel therapy for hemophilia B. Blood. 2010;115:4678-88.
92. Sabatino DE, Lange AM, Altynova ES, et al. Efficacy and safety of long-term prophylaxis in severe hemophilia A dogs following liver gene therapy using AAV vectors. Mol Ther. 2011;19:442-9.
93. Finn JD, Nichols TC, Svoronos N, et al. The efficacy and the risk of immunogenicity of FIX Padua (R338L) in hemophilia B dogs treated by AAV muscle gene therapy. Blood. 2012;120:4521-3.
94. Batty P, Mo AM, Hurlbut D, et al. Long-term follow-up of liver-directed, adeno-associated vector-mediated gene therapy in the canine model of hemophilia A. Blood. 2022;140:2672-83.
95. Suber YB, Doshi BS, French RA, Merricks E, Nichols T, Samelson-Jones B. Success of immune tolerance induction after AAV gene therapy in high-responding inhibitor hemophilia a dogs may be F8 genotype dependent. Blood. 2024;144:3579.
96. Carcao M, Königs C, Andersson NG, et al. Predictors of immune tolerance induction success in 231 children with severe hemophilia A with high-titer inhibitors - lessons learned from the PedNet prospective cohort study. J Thromb Haemost. 2025;23:3134-47.
97. Hay CR, DiMichele DM; International Immune Tolerance Study. The principal results of the International Immune Tolerance Study: a randomized dose comparison. Blood. 2012;119:1335-44.
98. Oomen I, Abdi A, Camelo RM, et al. Prediction of the chance of successful immune tolerance induction in persons with severe hemophilia A and inhibitors: a clinical prediction model. Res Pract Thromb Haemost. 2024;8:102580.
99. Zuccherato LW, Souza RP, Camelo RM, et al. Large deletions and small insertions and deletions in the factor VIII gene predict unfavorable immune tolerance induction outcome in people with severe hemophilia A and high-responding inhibitors. Thromb Res. 2024;242:109115.
100. Chitlur M, Warrier I, Rajpurkar M, Lusher JM. Inhibitors in factor IX deficiency a report of the ISTH-SSC international FIX inhibitor registry (1997-2006). Haemophilia. 2009;15:1027-31.
101. Scallan CD, Jiang H, Liu T, et al. Human immunoglobulin inhibits liver transduction by AAV vectors at low AAV2 neutralizing titers in SCID mice. Blood. 2006;107:1810-7.
102. Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342-7.
103. Boutin S, Monteilhet V, Veron P, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21:704-12.
104. Nichols T, Merricks E, Rakhe S, Samelson-Jones BJ, Pittman DD. OC69.3 Safe multi-year bleeding reduction by AAV gene therapy in neonatal and juvenile hemophilia B dogs. Available from: https://academy.isth.org/isth/2025/isth-2025-congress/4173051/tim.nichols.oc.69.3.-.safe.multi-year.bleeding.reduction.by.aav.gene.therapy.html [Last accessed on 25 Feb 2026].
105. Nguyen GN, Everett JK, Kafle S, et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat Biotechnol. 2021;39:47-55.
106. Keeler AM, Zhan W, Ram S, Fitzgerald KA, Gao G. The curious case of AAV immunology. Mol Ther. 2025;33:1946-65.
107. Li H, Lin SW, Giles-Davis W, et al. A preclinical animal model to assess the effect of pre-existing immunity on AAV-mediated gene transfer. Mol Ther. 2009;17:1215-24.
108. Buchlis G, Odorizzi P, Soto PC, et al. Enhanced T cell function in a mouse model of human glycosylation. J Immunol. 2013;191:228-37.
109. Schuettrumpf J, Liu JH, Couto LB, et al. Inadvertent germline transmission of AAV2 vector: findings in a rabbit model correlate with those in a human clinical trial. Mol Ther. 2006;13:1064-73.
110. Krause F, Schmidtke K, de Vasconcelos MF, et al. A shedding analysis after AAV8 CNS injection revealed fragmented viral DNA without evidence of functional AAV particles in mice. Gene Ther. 2024;31:345-51.
111. Highlights of prescribing information; 2023. Available from: https://www.novartis.com/us-en/sites/novartis_us/files/zolgensma.pdf [Last accessed on 25 Feb 2026].
112. Fewell JG, MacLaughlin F, Mehta V, et al. Gene therapy for the treatment of hemophilia B using PINC-formulated plasmid delivered to muscle with electroporation. Mol Ther. 2001;3:574-83.
113. Wang L, Nichols TC, Read MS, Bellinger DA, Verma IM. Sustained expression of therapeutic level of factor IX in hemophilia B dogs by AAV-mediated gene therapy in liver. Mol Ther. 2000;1:154-8.
114. Keeler GD, Markusic DM, Hoffman BE. Liver induced transgene tolerance with AAV vectors. Cell Immunol. 2019;342:103728.
115. Kumar SRP, Hoffman BE, Terhorst C, de Jong YP, Herzog RW. The Balance between CD8+ T cell-mediated clearance of AAV-encoded antigen in the liver and tolerance is dependent on the vector dose. Mol Ther. 2017;25:880-91.
116. Mingozzi F, Hasbrouck NC, Basner-Tschakarjan E, et al. Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. Blood. 2007;110:2334-41.
117. Samelson-Jones BJ, Arruda VR. Translational potential of immune tolerance induction by AAV liver-directed factor VIII gene therapy for hemophilia A. Front Immunol. 2020;11:618.
118. Valentino LA, Ozelo MC, Herzog RW, et al. A review of the rationale for gene therapy for hemophilia A with inhibitors: one-shot tolerance and treatment? J Thromb Haemost. 2023;21:3033-44.
119. Kaczmarek R, Samelson-Jones BJ, Herzog RW. Immune tolerance induction by hepatic gene transfer: first-in-human evidence. Mol Ther. 2024;32:863-4.
120. Young G. Induction of factor VIII tolerance by hemophilia gene transfer to eradicate factor VIII inhibitors. Blood Adv. 2025;9:265-9.
121. Samelson-Jones BJ, Doshi BS, George LA. Coagulation factor VIII: biological basis of emerging hemophilia A therapies. Blood. 2024;144:2185-97.
122. Simioni P, Tormene D, Tognin G, et al. X-linked thrombophilia with a mutant factor IX (factor IX Padua). N Engl J Med. 2009;361:1671-5.
123. Von Drygalski A, Giermasz A, Castaman G, et al. Etranacogene dezaparvovec (AMT-061 phase 2b): normal/near normal FIX activity and bleed cessation in hemophilia B. Blood Adv. 2019;3:3241-7.
124. Xue F, Li H, Wu X, et al. Safety and activity of an engineered, liver-tropic adeno-associated virus vector expressing a hyperactive Padua factor IX administered with prophylactic glucocorticoids in patients with haemophilia B: a single-centre, single-arm, phase 1, pilot trial. Lancet Haematol. 2022;9:e504-13.
125. Cheung AK, Hoggan MD, Hauswirth WW, Berns KI. Integration of the adeno-associated virus genome into cellular DNA in latently infected human Detroit 6 cells. J Virol. 1980;33:739-48.
126. Mccarty DM, Young SM, Samulski RJ. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu Rev Genet. 2004;38:819-45.
127. Morfopoulou S, Buddle S, Torres Montaguth OE, et al. Genomic investigations of unexplained acute hepatitis in children. Nature. 2023;617:564-73.
128. Servellita V, Sotomayor Gonzalez A, Lamson DM, et al. Adeno-associated virus type 2 in US children with acute severe hepatitis. Nature. 2023;617:574-80.
129. Martins KM, Breton C, Zheng Q, et al. Prevalent and disseminated recombinant and wild-type adeno-associated virus integration in macaques and humans. Hum Gene Ther. 2023;34:1081-94.
130. Bayard Q, Meunier L, Peneau C, et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat Commun. 2018;9:5235.
131. Fujimoto A, Furuta M, Totoki Y, et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016;48:500-9.
132. Nault J, Datta S, Imbeaud S, et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat Genet. 2015;47:1187-93.
133. Kaeppel C, Beattie SG, Fronza R, et al. A largely random AAV integration profile after LPLD gene therapy. Nat Med. 2013;19:889-91.
134. Sabatino DE, Bushman FD, Chandler RJ, et al. Evaluating the state of the science for adeno-associated virus integration: an integrated perspective. Mol Ther. 2022;30:2646-63.
135. Konkle BA, Walsh CE, Escobar MA, et al. BAX 335 hemophilia B gene therapy clinical trial results: potential impact of CpG sequences on gene expression. Blood. 2021;137:763-74.
136. Reiss UM, Davidoff AM, Tuddenham EG, et al. Sustained clinical benefit of AAV gene therapy in severe hemophilia B. N Engl J Med. 2025;392:2226-34.
137. Schmidt M, Foster GR, Coppens M, et al. Molecular evaluation and vector integration analysis of HCC complicating AAV gene therapy for hemophilia B. Blood Adv. 2023;7:4966-9.
138. Van Gorder L, Doshi BS, Willis E, et al. Analysis of vector genome integrations in multicentric lymphoma after AAV gene therapy in a severe hemophilia A dog. Mol Ther Methods Clin Dev. 2023;31:101159.
139. Batty P, Fong S, Franco M, et al. Vector integration and fate in the hemophilia dog liver multiple years after AAV-FVIII gene transfer. Blood. 2024;143:2373-85.
140. Chai Z, Zhang X, Dobbins AL, et al. Dexamethasone transiently enhances transgene expression in the liver when administered at late-phase post long-term adeno-associated virus transduction. Hum Gene Ther. 2022;33:119-30.
141. Jin X, Wu X, Song J, et al. Comparative evaluation of liver-directed knockin strategies with viral and nonviral vectors in mouse inherited disease models. Mol Ther. 2025;10:S1525-0016(25)01035-4.
142. Sarangi P, Kumar N, Sambasivan R, et al. AAV mediated genome engineering with a bypass coagulation factor alleviates the bleeding phenotype in a murine model of hemophilia B. Thromb Res. 2024;238:151-60.
143. Zhao J, Tian S, Peng Z, et al. Biomembrane-inspired lipid nanoparticles enhance CRISPR-Cas9 editing for hemophilia A. J Controlled Release. 2025;386:114141.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.