


Advances in the genomic and metabolic landscapes of acute myeloid leukemia
 0
0 
INTRODUCTION
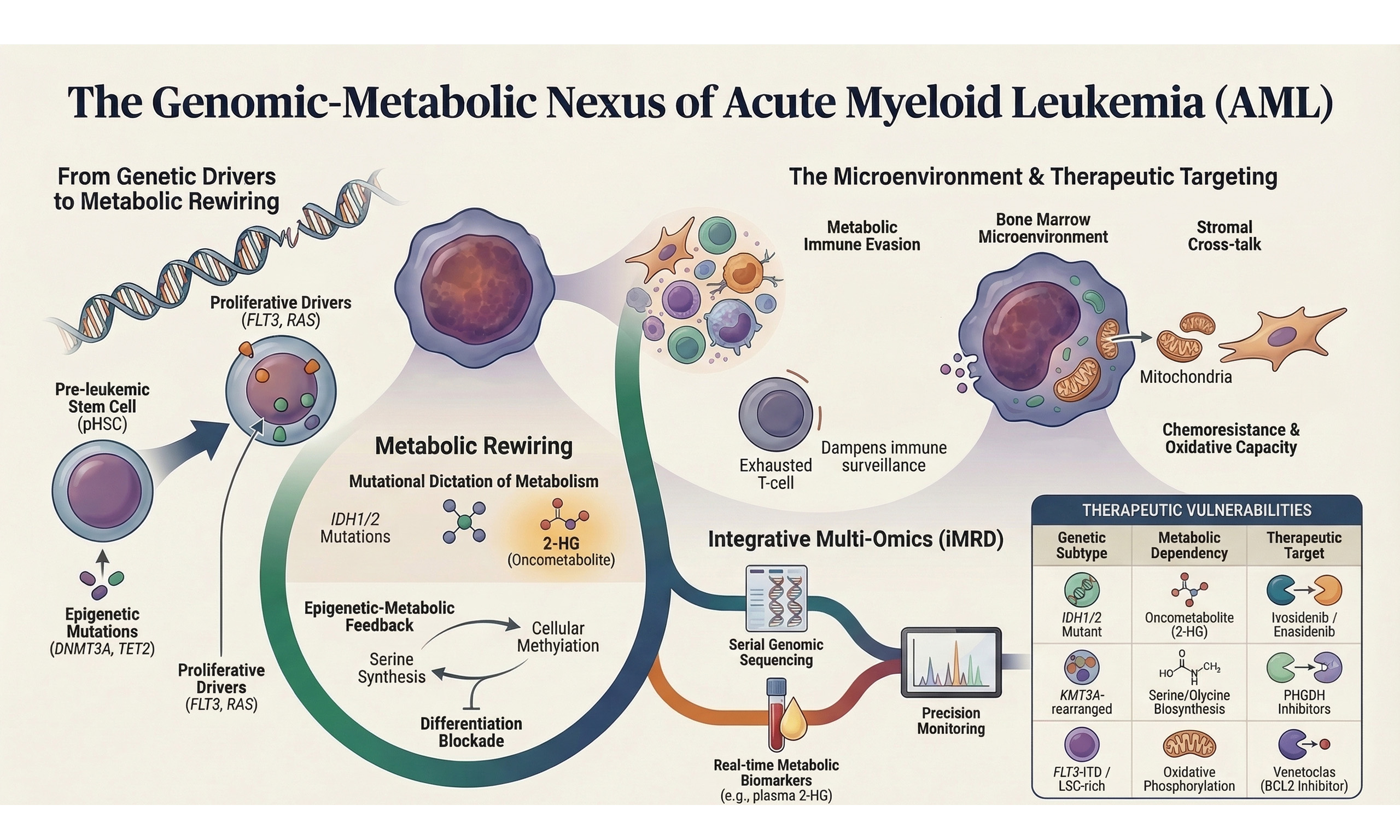
Acute myeloid leukemia (AML) represents the prototypical stem cell malignancy in which genetic lesions, epigenetic remodeling, and metabolic plasticity cooperate to subvert normal hematopoiesis. Despite incremental improvements brought by intensive chemotherapy, hematopoietic stem cell transplantation (HSCT), and more recently targeted agents, 5-year overall survival in adults remains approximately 35%[1]. The translational revolution of the past decade, encompassing whole-genome/exome sequencing (WGS/WES), single-cell transcriptomics, chromatin profiling, high-coverage metabolomics, and spatially resolved multi-omics, has revealed that leukemogenesis is driven not simply by a discrete set of driver mutations, but by a multidimensional network of metabolic, epigenetic, and microenvironmental alterations that confer fitness advantages to evolving leukemic clones[2,3]. Crucially, metabolic rewiring is not a passive by-product of oncogenic signaling but an active, hierarchically organized contributor to lineage fate determination, immune evasion, drug resistance, and relapse - a principle that carries direct implications for precision diagnostics and therapeutic targeting[3,4].
Recent comprehensive genomic studies have expanded the mutational landscape, identifying over 100 recurrently mutated genes distributed across critical pathways including signal transduction, epigenetic regulation, spliceosome machinery, and chromosomal cohesion complexes[5-7]. Concurrently, metabolomic profiling has uncovered AML-specific dependencies on oxidative phosphorylation (OxPhos), one-carbon metabolism, and amino acid flux, which are frequently genetically encoded[8]. These advances have revealed a fundamental organizing principle: oncogenic genotype directly encodes metabolic state - exemplified by Isocitrate Dehydrogenase 1/2 (IDH1/2) mutations that redirect α-ketoglutarate toward the oncometabolite 2-hydroxyglutarate (2-HG), and by KMT2A rearrangements that upregulate serine synthesis to sustain S-adenosylmethionine (SAM)-dependent epigenetic programs - while the resulting metabolic landscape, in turn, actively shapes immune microenvironmental interactions and defines the therapeutic vulnerabilities of each AML subtype[9-12].
We propose that the diverse metabolic alterations observed in AML are best understood not as a catalogue of parallel, context-dependent vulnerabilities, but as a genotype-encoded hierarchical program operating across three interconnected tiers. At the first tier, founding genomic and epigenetic lesions - including IDH1/2 mutations, KMT2A rearrangements, FLT3-ITD, and EZH2 inactivation - directly encode the primary metabolic identity of each AML subtype, determining dependencies on OxPhos, one-carbon flux, pyrimidine synthesis, or antioxidant defense. At the second tier, these genotype-encoded metabolic states sculpt the immune microenvironment through metabolite-mediated mechanisms: 2-HG suppresses T-cell receptor signaling, kynurenine and arginine depletion drive lymphocyte exhaustion, and extracellular succinate reprograms macrophages toward an immunosuppressive phenotype - collectively enabling leukemic immune evasion[13,14]. Critically, the microenvironment operates not as a passive recipient but as an active modulator at the third tier: hypoxia stabilizes hypoxia-inducible factor 1-alpha (HIF-1α) and reshapes mitochondrial metabolism, nutrient competition amplifies immunosuppressive metabolite gradients, and mesenchymal stromal cells restore oxidative capacity in therapy-stressed blasts through mitochondrial transfer - completing a self-reinforcing circuit that sustains clonal fitness and drives therapeutic resistance[15]. This perspective reviews current advances in the genomic and metabolic landscapes of AML through this hierarchical lens, with the dual aim of identifying core metabolic bottlenecks amenable to rational combination targeting and proposing a framework for integrative precision diagnostics that moves beyond static genotyping toward adaptive, multi-dimensional disease monitoring.
GENOMIC ARCHITECTURE AND BRANCHED CLONAL EVOLUTION
Comprehensive WGS/WES analyses encompassing over 5,000 annotated AML genomes now support an 11-13 group molecular taxonomy in which founding lesions - including nucleophosmin 1 (NPM1) mutations, RUNX1-RUNX1T1 translocations, core-binding factor beta-myosin heavy chain 11 (CBFB-MYH11) fusions, KMT2A rearrangements, tumor protein p53 (TP53) mutations with aneuploidy, and splicing/cohesin gene cluster alterations - define transcriptionally and metabolically distinct disease states[5-7]. Single-cell DNA and RNA sequencing, coupled with sophisticated phylogenetic reconstruction algorithms, have refined this taxonomy by demonstrating that leukemogenesis typically proceeds through a pre-leukemic hematopoietic stem cell (pHSC) stage[16,17]. These pHSCs accumulate age-related or therapy-related mutations predominantly in epigenetic modifier genes including DNA methyltransferase 3 alpha (DNMT3A), ten-eleven translocation 2 (TET2), additional sex combs-like 1 (ASXL1), and lysine methyltransferase 2D (KMT2D), which confer subtle self-renewal advantages while maintaining multilineage differentiation capacity - a phenomenon termed clonal hematopoiesis (CH)[8,18,19]. Subsequent acquisition of growth-factor receptor mutations {such as Fms-like tyrosine kinase 3 internal tandem duplication [FLT3-ITD] or tyrosine kinase domain [TKD] mutations, KIT Asp816 substitutions, and RAS (rat sarcoma viral oncogene homolog) pathway variants} or transcription factor lesions (including RUNX1 and CCAAT enhancer-binding protein alpha/CEBPA biallelic mutations) drives overt leukemic transformation and blast expansion[16,20].
Longitudinal sampling studies involving over 300 diagnosis-remission-relapse patient trios have revealed stereotypic evolutionary patterns with profound therapeutic implications[21-23]. These include: (i) linear evolution whereby a dominant founder clone persists through apparent remission and subsequently acquires resistance-conferring mutations that drive relapse; (ii) branching evolution in which a minor subclone harboring pre-existing resistance alleles preferentially expands under selective therapeutic pressure; and (iii) de novo clonal emergence from residual pHSCs carrying distinct founding mutations[21-23]. Such insights have catalyzed a paradigm shift from static one-time genotyping to adaptive genomic monitoring strategies. Current European LeukemiaNet (ELN) 2022 guidelines now recommend serial next-generation sequencing (NGS) at diagnosis and relapse for minimal residual disease (MRD) assessment, facilitating early detection of resistance-conferring alleles (exemplified by FLT3 Phe691Leu and BCL2 Gly101Val) and refinement of outcome prediction[1,24-26]. Recent validation studies conducted in 2024-2025 have confirmed the prognostic utility of ELN 2022 risk stratification in post-allogeneic HSCT settings, consistently demonstrating adverse outcomes in TP53-mutated and complex karyotype cases [Figure 1].
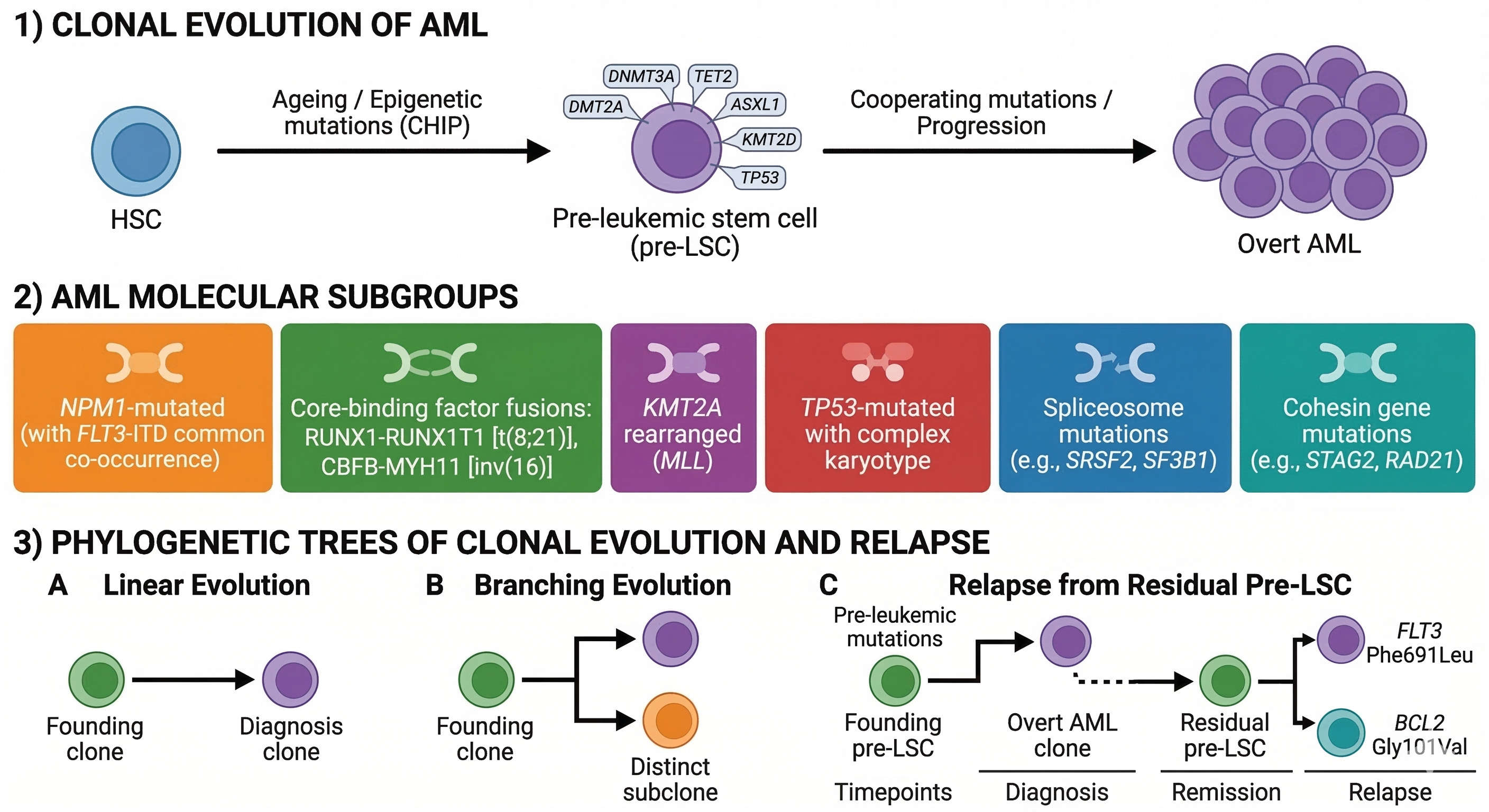
Figure 1. Genomic architecture and clonal evolution of AML. Schematic illustrating the progression from normal hematopoietic stem cells (HSCs) to pre-leukemic HSCs (pHSCs) via clonal hematopoiesis (CH) mutations (DNMT3A, TET2, ASXL1, KMT2D, TP53), followed by overt AML defined by 11-13 molecular subgroups (e.g., NPM1 mutations, RUNX1-RUNX1T1, CBFB-MYH11, KMT2A rearrangements, TP53/aneuploidy, spliceosome/cohesin clusters). Right panel depicts archetypal evolutionary patterns (linear, branching, de novo from pHSCs) with resistance mutations (e.g., FLT3 Phe691Leu, BCL2 Gly101Val) and serial NGS monitoring timepoints per ELN 2022 guidelines. Figure generated using AI-based tools and finalized by the authors in Microsoft PowerPoint. AML: Acute myeloid leukemia; NGS: next-generation sequencing; AI: artificial intelligence.
EPIGENETIC REGULATORS AND THEIR METABOLIC FOOTPRINTS
Beyond signal transduction drivers, mutational inactivation or neomorphic activation of epigenetic regulatory proteins contributes substantially to well-defined metabolic footprints that characterize specific AML subsets[9,27,28]. The paradigmatic example remains IDH1 Arg132 and IDH2 Arg140/172 mutations, which confer neomorphic reductive enzymatic activity that converts α-ketoglutarate (α-KG) to the (R)-enantiomer of 2-hydroxyglutarate[9,10]. This oncometabolite competitively inhibits multiple α-KG-dependent dioxygenases including TET DNA demethylases, Jumonji domain-containing histone demethylases, and prolyl-hydroxylases, culminating in widespread promoter CpG island hypermethylation, differentiation arrest, and hypoxia-inducible factor 1-alpha (HIF1α) stabilization[29].
Metabolomic quantification of 2-HG in plasma and bone marrow aspirates serves as both a diagnostic biomarker and a pharmacodynamic readout for evaluating the efficacy of small-molecule IDH1/2 inhibitors[9,10,30,31]. Specifically, serial 2-HG measurements provide real-time pharmacodynamic monitoring for the selective inhibitors ivosidenib (targeting mutant IDH1) and enasidenib (targeting mutant IDH2)[9,10,30,31]. Across over 500 patients treated in phase I/II/III trials, overall complete remission plus complete remission with partial hematologic recovery (CR/CRh) rates approximated 35%-40% with median overall survival approaching 30 months - representing a remarkable therapeutic achievement in relapsed/refractory (R/R) AML[30-32]. Notably, while a 90% decline in on-treatment 2-HG levels by cycle 1 day 15 indicates successful on-target inhibitor activity, 2-HG suppression alone did not uniformly predict clinical response in enasidenib-treated patients. Most non-responding patients also exhibited substantial 2-HG suppression, suggesting that additional off-target mechanisms beyond direct IDH2 inhibition ultimately determine therapeutic efficacy[33].
Parallel metabolic-epigenetic paradigms are emerging in AML characterized by mutations in enhancer of zeste homolog 2 (EZH2), BCL6 corepressor (BCOR), and SET domain containing 2 (SETD2). Loss-of-function EZH2 mutations deplete H3K27me3 repressive marks in myeloid progenitors, potentially creating compensatory dependencies on methionine cycle flux to sustain residual cellular methylation capacity[34]. Emerging evidence from related myeloid and lymphoid malignancies suggests that such epigenetic insufficiency generates synthetic-lethal interactions with constraints on one-carbon metabolism - including MAT2A-mediated S-adenosylmethionine production - a mechanistic paradigm that warrants prospective investigation in EZH2-mutant AML as a candidate metabolic vulnerability[11]. Recent 2024 analyses have further linked TP53-associated AML hallmarks to profoundly altered cellular metabolism mediated through epigenetic dysregulation mechanisms [Figure 2].
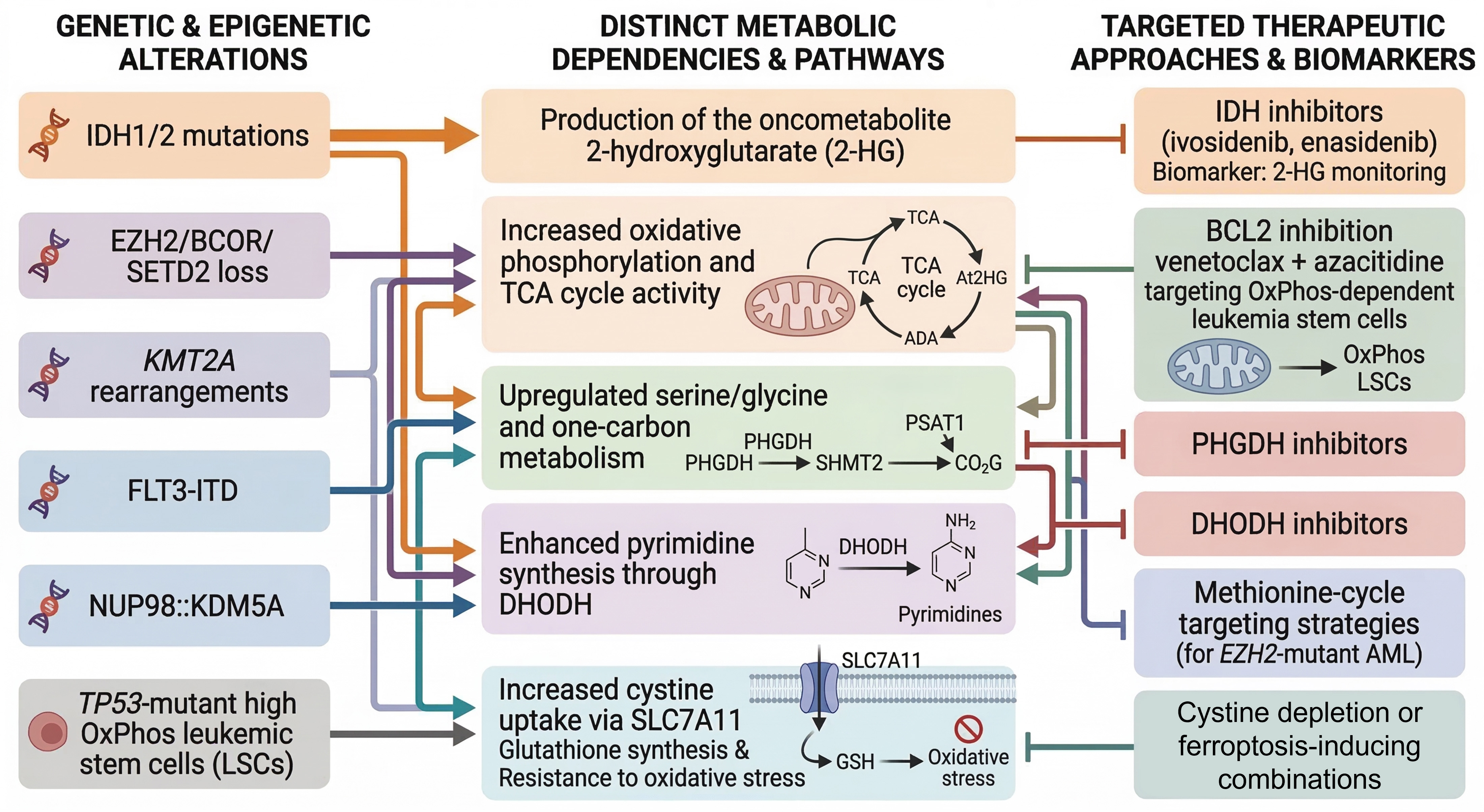
Figure 2. Genotype-metabolism-therapy axis in AML. Mechanistic diagram linking key genetic/epigenetic drivers (IDH1/2, EZH2/BCOR/SETD2 loss, KMT2A-r/FLT3-ITD/NUP98::KDM5A, TP53-mutant OxPhos-high LSCs) to metabolic hallmarks (2-HG production, serine/glycine/one-carbon flux via PHGDH/PSAT1/SHMT2, pyrimidine synthesis via DHODH, cystine uptake via SLC7A11). Targeted therapies and biomarkers include IDH inhibitors (ivosidenib/enasidenib) with 2-HG monitoring, venetoclax/azacitidine for OxPhos LSCs, PHGDH/DHODH inhibition, and SLC7A11/cyst(e)inase-mediated ferroptosis induction Figure generated using AI-based tools and finalized by the authors in Microsoft PowerPoint. AML: Acute myeloid leukemia; TCA: tricarboxylic acid; PHGDH phosphoglycerate dehydrogenase; PSAT1: phosphoserine aminotransferase; SHMT2: serine hydroxymethyltransferase 2; DHODH: dihydroorotate dehydrogenase; ADA: adenosine deaminase; IDH: isocitrate dehydrogenase; AI: artificial intelligence; DHODH: dihydroorotate dehydrogenase.
METABOLIC HALLMARKS BEYOND ONCOMETABOLITES AND THEIR GENETIC DETERMINANTS
Multiple studies have identified metabolic hallmarks extending well beyond oncometabolite accumulation. Leukemia-initiating cells (LICs), prospectively isolated using the CD34+CD38-CD99hi immunophenotype, exhibit elevated mitochondrial membrane potential (ΔΨm), increased steady-state tricarboxylic acid (TCA) cycle intermediate concentrations, and robust spare respiratory capacity - collectively providing substantial biosynthetic and energetic advantages[9,35-37]. Genome-wide CRISPR-Cas9 knockout screens have pinpointed electron transport chain (ETC) complex II components (succinate dehydrogenase subunits SDHA/B) and mitochondrial ribosomal proteins (MRPS12, MRPL45) as non-redundant metabolic dependencies in AML[38]. The BCL2 inhibitor venetoclax exerts its anti-leukemic stem cell (anti-LSC) effects partially through displacement of BCL2-mediated sequestration of the pro-apoptotic protein BAX (BCL2 Associated X, Apoptosis Regulator), thereby triggering mitochondrial outer membrane permeabilization (MOMP) and subsequent cytochrome c release - an effect particularly potentiated in OxPhos-dependent leukemic cell populations[39,40].
Specific AML genetic subtypes may harbor genotype-encoded dependencies on de novo serine/glycine biosynthesis, linking oncogenic drivers such as FLT3-ITD, NUP98::KDM5A fusions, and KMT2A rearrangements to upregulation of the serine synthesis pathway, including phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase 1 (PSAT1), and serine hydroxymethyltransferase 2 (SHMT2)[11,41]. Critically, serine-derived one-carbon units sustain S-adenosylmethionine (SAM) production, thereby fueling histone and DNA methylation and potentially reinforcing the epigenetic blockade of myeloid differentiation - positioning PHGDH and the broader one-carbon pathway as candidate therapeutic targets in molecularly defined AML subsets that warrant prospective validation in primary patient material[11,12,42]. The Activating Transcription Factor 4 (ATF4)/PSAT1/c-Jun N-terminal kinase (JNK) signaling axis has recently been identified as a ferroptosis suppressor driving venetoclax resistance in AML, further underscoring the cross-sectional therapeutic relevance of serine pathway targeting[43].
Alternatively, pyrimidine biosynthesis has been causally linked to differentiation blockade in specific AML contexts[36,41]. Dihydroorotate dehydrogenase (DHODH) couples de novo pyrimidine synthesis to ETC complex III activity, thereby establishing a critical biosynthetic-bioenergetic nexus[41]. DHODH inhibitors synergize with differentiation-inducing agents such as all-trans retinoic acid (ATRA) in preclinical AML models, and pyrimidine biosynthetic stress modulates differentiation responses through ribonucleotide reductase-dependent mechanisms, revealing context-dependent metabolic vulnerabilities amenable to combination therapeutic targeting[41,44]. Nucleotide metabolism and epigenetic regulation have furthermore been identified as cooperative determinants of the differentiation blockade that defines AML pathobiology[45].
AML blasts experience chronic oxidative stress resulting from aberrant Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, elevated ETC-mediated reactive oxygen species (ROS) production, and residence within cytokine-rich bone marrow microenvironmental niches. Consequently, many AML subtypes display marked dependence on cystine uptake mediated by the system xc- (SLC7A11) antiporter, which imports extracellular cystine for subsequent reduction to cysteine and incorporation into glutathione for antioxidant defense[46,47]. Pharmacologic or genetic inhibition of SLC7A11, or exposure to cystine-depleting enzymes [cyst(e)inase], induces lethal lipid peroxidation. When combined with agents that disrupt mitochondrial electron flux, leukemic cells accumulate toxic dihydrolipoyl species and become primed for ferroptotic cell death[47-49]. A ketogenesis-ferroptosis axis has been identified as maintaining leukemic stem cell survival and promoting disease progression, establishing lipid metabolic reprogramming as a novel node of ferroptosis resistance in the LSC compartment[50]. PIEZO1 has been shown to sustain AML progression and LSC maintenance through the HIF1A-SLC7A11 axis, providing a mechanistic link between hypoxia signaling and ferroptosis defense in AML stem cells[51]. Recent comprehensive analyses have identified metabolism-related gene signatures including CLIC2, CA13, and KCNJ2 as prognostically significant factors modulating cysteine and methionine metabolic pathways in AML. A critical re-evaluation of the rationale for targeting OxPhos in AML highlights the context-dependency of this vulnerability and cautions against uniform extrapolation across genetically heterogeneous patient populations[52].
METABOLIC IMMUNE EVASION AND MICROENVIRONMENTAL CROSS-TALK
Aberrant metabolic profiles in AML have been directly correlated with immune evasion mechanisms - and, critically, these immunomodulatory effects are not generic but genotype-specific: IDH1/2-mutant blasts generate extracellular 2-HG that directly suppresses T-cell receptor signaling; FLT3-ITD-driven Signal Transducer and Activator of Transcription 5 (STAT5) hyperactivation transcriptionally induces IDO1 expression; and TP53-mutant AML remodels the immune microenvironment toward a profoundly immunosuppressive configuration through coordinated metabolic and non-metabolic mechanisms[27,53]. Transcriptomic meta-analysis of 1,002 diagnostic AML samples identified recurrent upregulation of arginase-2 (ARG2) and indoleamine 2,3-dioxygenase 1 (IDO1) - enzymes that catabolize essential amino acids arginine and tryptophan into immunosuppressive metabolites ornithine and kynurenine, respectively[13]. Kynurenine accumulation within the leukemic bone marrow microenvironment has been proposed to contribute to T-cell exhaustion, including expansion of PD-1+TIM-3+ subsets, and may carry adverse prognostic implications - although prospective clinical validation in molecularly defined AML cohorts remains an unmet need[13,53]. These observations establish the ARG2/IDO1 metabolic-immune axis as a mechanistically compelling therapeutic target, particularly in combination with venetoclax-based regimens, warranting rigorous evaluation in early-phase clinical trials.
These metabolic-immune interactions extend to adoptive cellular therapies. Chimeric antigen receptor (CAR) T-cell exhaustion within the bone marrow microenvironment is driven by nutrient deprivation, hypoxia, and accumulation of immunosuppressive metabolites. Gene editing strategies to overexpress peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) or knock out programmed cell death protein 1 (PD-1) improve mitochondrial fitness and enhance anti-leukemic efficacy in preclinical models[54].
Furthermore, bone marrow mesenchymal stromal cells (MSCs) protect leukemic progenitor cells by donating functional mitochondria through connexin-43-dependent tunneling nanotubes (TNTs), thereby conferring chemoresistance and restoring oxidative metabolic capacity in therapy-stressed AML cells[15]. Pharmacologic blockade of mitochondrial transfer or direct inhibition of OxPhos in recipient blast cells can partially re-sensitize leukemia to conventional cytarabine-based chemotherapy. These observations illustrate a critical principle: the bone marrow microenvironment is not merely a passive recipient of blast-derived metabolic signals, but functions as an active, independent modulator of leukemic cell metabolism - reshaping energetic dependencies, drug sensitivity profiles, and clonal evolutionary trajectory in ways not captured by genomic characterization of bulk tumor cells alone[55].
Leukemic cells actively secrete metabolites including lactate, succinate, and 2-HG, each exerting distinct effects on stromal and immune effector cell populations. Extracellular succinate engages the G-protein-coupled receptor SUCNR1 on macrophages, driving an immunosuppressive M2-like phenotype characterized by high interleukin-10 (IL-10) and low tumor necrosis factor-alpha (TNF-α) secretion. Diffusible 2-HG enters T cells where it inhibits lymphocyte-specific protein tyrosine kinase (LCK) phosphorylation and dampens T-cell receptor (TCR) signaling, thereby contributing to suboptimal performance of both native and CAR-modified T cells within the leukemic bone marrow microenvironment[14]. Beyond these direct metabolite-mediated effects, hypoxic niches within the leukemic marrow stabilize HIF-1α in blast populations, driving transcriptional upregulation of PD-L1 and amplifying immune checkpoint-mediated T-cell suppression - while simultaneously feeding back to remodel leukemic cell metabolism by promoting glycolytic flux, suppressing mitochondrial biogenesis, and enhancing OxPhos dependency in residual stem-like populations[29,53]. This bidirectionality - whereby leukemic metabolic states shape immune function, and microenvironmental signals in turn reconfigure leukemic metabolism - exemplifies the self-reinforcing circuit proposed as the organizing principle of this perspective, and identifies the convergence of oncometabolite signaling with immune checkpoint biology as a mechanistically compelling frontier for combinatorial therapeutic targeting [Figure 3].
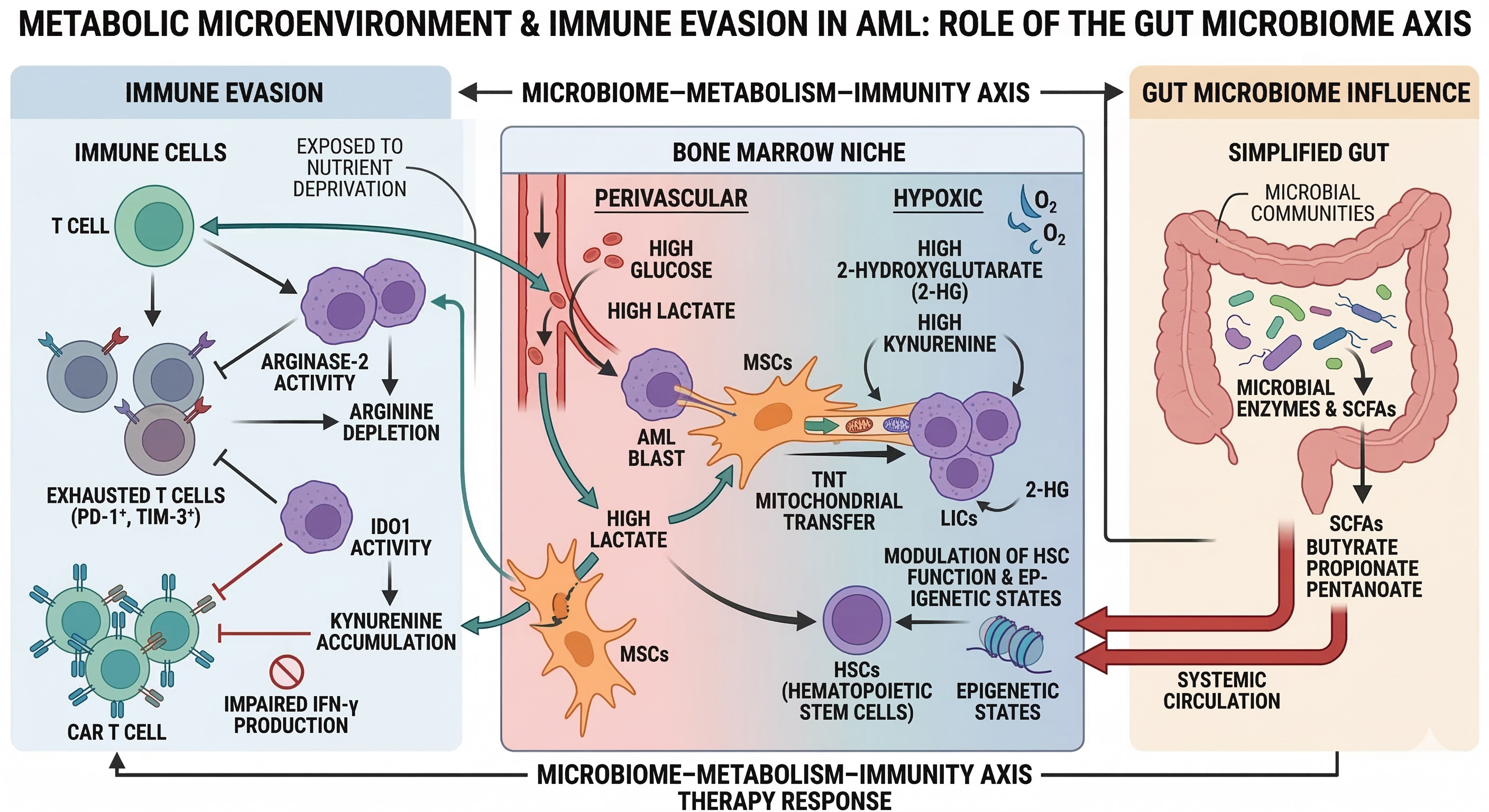
Figure 3. Metabolic microenvironment, immune evasion, and microbiome axis in AML. Bone marrow niche model showing AML blasts/LICs interacting with stromal cells via mitochondrial-transferring tunneling nanotubes, spatially heterogeneous metabolites (glucose/lactate perivascularly; 2-HG/kynurenine hypoxic zones), and immunosuppressive pathways (ARG2/IDO1 depleting arginine/tryptophan, impairing PD-1+TIM-3+ T cells and CAR-T). Gut microbiome-derived short-chain fatty acids (SCFAs; butyrate/propionate) modulate systemic HSCs, epigenetics, and therapy responses. Figure generated using AI-based tools and finalized by the authors in Microsoft PowerPoint. AML: Acute myeloid leukemia; LICs: leukemia-initiating cells; HSCs: hematopoietic stem cells; CAR-T: chimeric antigen receptor-T; AI: artificial intelligence.
Recent evidence suggests that systemic metabolites generated by the gut microbiome, particularly short-chain fatty acids (SCFAs) such as butyrate and propionate, can modulate hematopoietic stem cell function and influence AML progression[56,57]. Dysbiosis in AML patients has been associated with
CLINICAL TRANSLATION AND INTEGRATIVE DIAGNOSTICS
One of the major challenges confronting contemporary clinical practice involves developing practical diagnostic platforms that simultaneously capture both genetic and metabolic profiling data. The Beat-AML master trial pioneered integration of ex vivo high-throughput drug sensitivity testing with comprehensive genomic profiling to facilitate rational matching of patients to pathway-targeted therapeutic agents[58]. Despite successfully demonstrating technical feasibility, composite clinical response rates did not significantly exceed historical controls, underscoring the critical necessity of layering detailed metabolic readouts onto genetic risk stratification schemes[58]. In this context, current ELN 2022 risk stratification - while representing the established clinical standard for genomic risk classification at diagnosis - provides a static, single-timepoint assessment that does not capture metabolic state, immunological evolution, or the dynamic clonal trajectories that emerge under therapeutic pressure[1]. Integrating metabolic readouts into ELN-based frameworks represents a conceptually compelling approach to address this inherent limitation.
Integrative MRD (iMRD) approaches aim to merge longitudinal variant-allele-frequency (VAF) trajectories - the cornerstone of ELN 2022-endorsed NGS-based MRD monitoring at diagnosis, post-induction, and pre-transplant timepoints - with serial metabolic biomarker measurements, potentially enhancing sensitivity and lead time of relapse prediction beyond what genomic surveillance alone achieves[1,59,60]. The most clinically mature example is serial monitoring of plasma or serum 2-HG in IDH-mutant AML treated with ivosidenib or enasidenib: a sustained ≥ 90% decline by cycle 1, day 15 confirms on-target enzymatic inhibition, while subsequent 2-HG re-elevation may signal pharmacodynamic failure or emergence of resistance mechanisms prior to morphologic relapse, enabling earlier therapeutic reassessment than conventional criteria permit[9,10,60]. Concordant molecular and metabolic MRD negativity - undetectable VAF by sensitive NGS alongside suppressed 2-HG - represents the strongest available evidence of deep remission and may ultimately inform transplant timing decisions in eligible patients, whereas discordant results warrant intensified monitoring and mechanistic investigation. Beyond IDH-mutant disease, plasma profiling of arginine and kynurenine may provide complementary insight into residual immunosuppressive disease burden, as supported by metabolomics profiling studies identifying systemic metabolic differences associated with drug sensitivity in primary AML[13,61]. This approach currently lacks analytical standardization and prospective clinical validation required for routine practice and should be considered an investigational endpoint within structured trial frameworks.
One of the most intriguing technological approaches under active development is spatial metabolomics. Matrix-assisted laser desorption/ionization (MALDI) imaging mass spectrometry (IMS) enables in situ mapping of metabolites, lipids, and small molecules across intact tissue sections with subcellular spatial resolution[62,63]. While initial applications of MALDI-IMS have demonstrated spatially segregated metabolic niches in solid tumor microenvironments - including coexisting perivascular glucose-enriched zones and hypoxic, nutrient-depleted regions - the systematic application of this technology to diagnostic AML bone marrow specimens remains a frontier of active investigation[62,63]. Translating MALDI-IMS to AML holds particular promise for revealing how genotype-encoded metabolic reprogramming generates spatially heterogeneous microenvironmental niches that may underlie differential therapeutic responses poorly captured by conventional bulk genomic profiling alone. Realizing this translational potential will require standardized tissue-processing protocols compatible with routine diagnostic bone marrow trephine biopsies, curated metabolite reference databases specific to hematologic malignancies, and prospective studies correlating MALDI-IMS-derived spatial signatures with genomic risk groups and clinical outcomes.
Finally, therapeutic manipulation of the gut microbiome through targeted dietary interventions, prebiotic supplementation, or fecal microbiota transplantation (FMT) represents an emerging frontier in supportive care and metabolic reprogramming strategies for AML. SCFA-mediated modulation of the metabolic-epigenetic crosstalk in hematopoietic cells may offer novel opportunities to influence disease trajectory and treatment outcomes[56,57,64].
Translating these integrative approaches into actionable clinical practice requires a staged implementation strategy that respects current evidence boundaries while establishing the infrastructure for progressive adoption. We propose a three-tier integrative diagnostic framework as a conceptual scaffold for future trial design and clinical pathway development. At the first tier, applicable to all AML patients at diagnosis, ELN 2022 genomic risk classification is complemented by baseline metabolomic profiling - including plasma 2-HG (not limited to cases with known IDH mutations at referral) and a standardized amino acid panel - establishing a metabolic baseline against which on-treatment changes can be tracked. At the second tier, applicable to patients receiving targeted therapies or enrolled in clinical trials, serial metabolic monitoring is integrated with NGS-MRD surveillance at ELN-recommended timepoints: 2-HG kinetics for IDH inhibitor-treated patients, and immune-metabolic markers for immunotherapy combination trials, enabling concordance analysis that refines remission depth assessment and early relapse detection. At the third tier, within academic and translational research frameworks, spatial metabolomics of bone marrow biopsies and longitudinal microbiome profiling are incorporated to characterize microenvironmental heterogeneity and systemic metabolic modulators. This framework is explicitly proposed as a progressive extension of - not a replacement for - established ELN 2022 standards, capturing dimensions of AML biology that current diagnostic paradigms do not address[1,58,61]. Proof of principle for metabolic targeting in hematologic malignancies has been established in related disease contexts[65]. AML molecular profiling data from geographically diverse populations suggest that metabolic dependencies may further vary across ethnic and geographic backgrounds, an important equity consideration for translating precision metabolic targeting[66].
CONCLUSION
In conclusion, AML exemplifies an exceptionally tight reciprocal relationship between oncogenic genotype and cellular metabolic state. Large-scale sequencing efforts have successfully stratified AML into molecularly coherent subgroups, each characterized by distinctive metabolic dependencies and immunologic vulnerabilities that can be therapeutically exploited. The clinical translation of these fundamental insights hinges critically on development and implementation of integrative diagnostic platforms that seamlessly combine genomic surveillance, comprehensive metabolomic profiling, spatial analytical techniques, and microbiome characterization. Such platforms hold substantial potential to enable adaptive therapeutic strategies that dynamically adjust treatment interventions in real-time based on evolving clonal landscapes and shifting metabolic states, ultimately improving clinical outcomes for patients confronting this challenging and heterogeneous disease.
DECLARATIONS
Authors’ contributions
Conceptualization, supervision: Piccaluga PP, Baran N
Resources, project administration, funding acquisition: Piccaluga PP
Data curation, writing - original draft preparation, writing - review and editing, methodology, investigation: Piccaluga PP, Tsekhovska V, Cimmino L, Lopiano M, Berisha A, Lapaj I, Visani G, Baran N
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool Gemini (version 2.5 Flash, released 2025-05-22) was used solely for figures conceptualization and editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript
Financial support and sponsorship
The work reported in this publication was funded by the Italian Ministry of Health, RC-2025-2797263 and by PRIN 2022NXK38S.
Conflicts of interest
Piccaluga PP is an Editorial Board Member of the Journal of Translational Genetics and Genomics. Piccaluga PP was not involved in any aspect of the editorial process for this manuscript, including reviewer selection, manuscript handling, and decision-making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140:1345-77.
2. Khattab S, Berisha A, Baran N, Piccaluga PP. Rat sarcoma virus family genes in acute myeloid leukemia: pathogenetic and clinical implications. Biomedicines. 2025;13:202.
3. Vosberg S, Greif PA. Clonal evolution of acute myeloid leukemia from diagnosis to relapse. Genes Chromosomes Cancer. 2019;58:839-49.
4. Kamath-Loeb AS, Shen JC, Schmitt MW, et al. Accurate detection of subclonal variants in paired diagnosis-relapse acute myeloid leukemia samples by next generation duplex sequencing. Leuk Res. 2022;115:106822.
5. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209-21.
6. Tyner JW, Tognon CE, Bottomly D, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562:526-31.
7. Haferlach T, Walter W. Challenging gold standard hematology diagnostics through the introduction of whole genome sequencing and artificial intelligence. Int J Lab Hematol. 2023;45:156-62.
8. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488-98.
9. Stein EM, DiNardo CD, Fathi AT, et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood. 2019;133:676-87.
10. DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378:2386-98.
11. Pikman Y, Puissant A, Alexe G, et al. Targeting MTHFD2 in acute myeloid leukemia. J Exp Med. 2016;213:1285-306.
12. Ye J, Fan J, Venneti S, et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014;4:1406-17.
13. Mussai F, De Santo C, Abu-Dayyeh I, et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood. 2013;122:749-58.
14. Böttcher M, Renner K, Berger R, et al. D-2-hydroxyglutarate interferes with HIF-1α stability skewing T-cell metabolism towards oxidative phosphorylation and impairing Th17 polarization. Oncoimmunology. 2018;7:e1445454.
15. Moschoi R, Imbert V, Nebout M, et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood. 2016;128:253-64.
16. Zeng AGX, Iacobucci I, Shah S, et al. Single-cell transcriptional atlas of human hematopoiesis reveals genetic and hierarchy-based determinants of aberrant AML differentiation. Blood Cancer Discov. 2025;6:307-24.
17. Hörst K, Kühn C, Haferlach C, Haferlach T, Khoury JD. Genetic studies in clonal haematopoiesis, myelodysplastic neoplasms and acute myeloid leukaemia- a practical guide to WHO-HAEM5. Med Genet. 2024;36:21-9.
18. Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477-87.
19. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9-16.
20. Pourebrahim R, Montoya RH, Akiyama H, et al. Age-specific induction of mutant p53 drives clonal hematopoiesis and acute myeloid leukemia in adult mice. Cell Rep Med. 2024;5:101558.
21. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506-10.
22. Shlush LI, Mitchell A, Heisler L, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature. 2017;547:104-8.
23. Morita K, Wang F, Jahn K, et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat Commun. 2020;11:5327.
24. Ruhnke L, Bill M, Zukunft S, et al. Validation of the revised 2022 European LeukemiaNet risk stratification in adult patients with acute myeloid leukemia. Blood Adv. 2025;9:1392-404.
25. Mrózek K, Kohlschmidt J, Blachly JS, et al. Outcome prediction by the 2022 European LeukemiaNet genetic-risk classification for adults with acute myeloid leukemia: an alliance study. Leukemia. 2023;37:788-98.
26. Lachowiez CA, Long N, Saultz J, et al. Comparison and validation of the 2022 European LeukemiaNet guidelines in acute myeloid leukemia. Blood Adv. 2023;7:1899-909.
27. Chomczyk M, Gazzola L, Dash S, et al. Impact of p53-associated acute myeloid leukemia hallmarks on metabolism and the immune environment. Front Pharmacol. 2024;15:1409210.
28. DiNardo CD, Tiong IS, Quaglieri A, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135:791-803.
29. Losman JA, Looper RE, Koivunen P, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621-5.
30. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722-31.
31. Roboz GJ, DiNardo CD, Stein EM, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood. 2020;135:463-71.
32. Montesinos P, Recher C, Vives S, et al. Ivosidenib and azacitidine in IDH1-mutated acute myeloid leukemia. N Engl J Med. 2022;386:1519-31.
33. Amatangelo MD, Quek L, Shih A, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017;130:732-41.
34. Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722-6.
35. Jones CL, Stevens BM, D’Alessandro A, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. 2018;34:724-740.e4.
36. Farge T, Saland E, de Toni F, et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017;7:716-35.
37. Lagadinou ED, Sach A, Callahan K, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329-41.
38. Wang T, Yu H, Hughes NW, et al. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic ras. Cell. 2017;168:890-903.e15.
39. Pollyea DA, Stevens BM, Jones CL, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24:1859-66.
40. Sharon D, Cathelin S, Mirali S, et al. Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Sci Transl Med. 2019;11:eaax2863.
41. Sykes DB, Kfoury YS, Mercier FE, et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell. 2016;167:171-186.e15.
42. Pacold ME, Brimacombe KR, Chan SH, et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol. 2016;12:452-8.
43. Zhu X, Huang W, Zhang M, Tao Y, Zhang H. The ATF4/PSAT1/JNK signaling axis suppresses ferroptosis to drive venetoclax resistance in AML. Exp Cell Res. 2026;460:115045.
44. Brcic A, Lalic H, Smoljo T, et al. Roles of RRM2 and RRM2B in pyrimidine stress responses and differentiation of acute myeloid leukemia cells. Cell Death Discov. 2026;12:271.
45. Takahashi S. Targeting nucleotide metabolism and epigenetic regulation to overcome the differentiation blockade in AML. Leuk Res Rep. 2026;25:100586.
46. Cramer SL, Saha A, Liu J, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med. 2017;23:120-7.
47. Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the xc- cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633-40.
48. Magtanong L, Ko PJ, To M, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. 2019;26:420-432.e9.
50. Han X, Wang K, Ma W, et al. A ketogenesis-ferroptosis axis maintains leukemic stem cell survival and leukemia progression. Cell Stem Cell. 2026;33:1016-1030.e6.
51. Zhang T, Cao Z, Gao K, et al. PIEZO1 is required for acute myeloid leukemia progression and leukemia stem cell maintenance via HIF1A-SLC7A11 axis-mediated ferroptosis defense. Adv Sci. 2026;e75648.
52. Chrest BR, Fisher-Wellman KH. Re-evaluating the rationale for targeting oxidative phosphorylation in acute myeloid leukemia. Biochem J. 2026;483:907-25.
53. Narote S, Desai SA, Patel VP, Deshmukh R, Raut N, Dapse S. Identification of new immune target and signaling for cancer immunotherapy. Cancer Genet. 2025;294-295:57-75.
54. Fraietta JA, Nobles CL, Sammons MA, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558:307-12.
55. Khattab S, El Sorady M, El-Ghandour A, Visani G, Piccaluga PP. Hematopoietic and leukemic stem cells homeostasis: the role of bone marrow niche. Explor Target Antitumor Ther. 2024;5:1027-55.
56. Luu M, Pautz S, Kohl V, et al. The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic-epigenetic crosstalk in lymphocytes. Nat Commun. 2019;10:760.
57. Payen VL, Mina E, Van Hée VF, Porporato PE, Sonveaux P. Monocarboxylate transporters in cancer. Mol Metab. 2020;33:48-66.
58. Bottomly D, Long N, Schultz AR, et al. Integrative analysis of drug response and clinical outcome in acute myeloid leukemia. Cancer Cell. 2022;40:850-864.e9.
59. Hourigan CS. Role of MRD testing in acute myeloid leukemias. What is the status and how to get there? Semin Hematol 2025;62:431-7.
60. de Botton S, Montesinos P, Schuh AC, et al. Enasidenib vs conventional care in older patients with late-stage mutant-IDH2 relapsed/refractory AML: a randomized phase 3 trial. Blood. 2023;141:156-67.
61. Brattås MK, Hatfield KJ, Paulsen Rye K, Reikvam H. Metabolomics profiling of acute myelogenous leukemia patients to identify systemic differences associated with in vitro sensitivity to SYK inhibitors. Metabolomics. 2026;22;51.
63. Schwamborn K, Caprioli RM. Molecular imaging by mass spectrometry-looking beyond classical histology. Nat Rev Cancer. 2010;10:639-46.
64. Galloway-Peña JR, Smith DP, Sahasrabhojane P, et al. The role of the gastrointestinal microbiome in infectious complications during induction chemotherapy for acute myeloid leukemia. Cancer. 2016;122:2186-96.
65. Bagaloni I, Visani A, Biagiotti S, et al. Metabolic switch and cytotoxic effect of metformin on burkitt lymphoma. Front Oncol. 2021;11:661102.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.