



Electrochemical biosensing across microsampled blood and interstitial fluid for metabolic, hormonal, and therapeutic monitoring
 0
0 
Abstract
Electrochemical biosensing is advancing beyond traditional single-analyte glucose monitoring toward platforms capable of quantifying metabolites, hormones, and therapeutic agents across microsampled blood and continuously accessed interstitial fluid (ISF). Progress in biorecognition chemistry, signal transduction, and skin-integrated interfaces has enabled minimally invasive access to capillary blood and ISF using microneedle-based and implantable devices. ISF-based sensing introduces depth-dependent analyte gradients, ISF-blood transport kinetics, and characteristic signal lag, necessitating sampling-aware sensor design and quantitative models for systemic correlation. We review device architectures that support stable, multi-day operation and outline calibration strategies for predicting blood-equivalent concentrations. Emerging directions include hybrid systems that merge electrochemical detection with complementary modalities, multi-analyte platforms that capture metabolic interdependence, and closed-loop approaches coupling biochemical readout with automated intervention. Together, these advances establish electrochemical biosensing as a core technology for continuous and clinically interpretable biochemical monitoring across blood and ISF.
Keywords
INTRODUCTION
The clinical success of glucose biosensors, from early blood glucose meters (BGMs) to modern continuous glucose monitors (CGMs), represents a paradigm shift in the electrochemical capture and quantification of biochemical data[1,2]. Built on enzymatic redox reactions, these systems transformed a fundamental biochemical reaction into a compact digital device capable of providing real-time, quantitative information[3]. Their reliability, affordability, and simplicity established electrochemical transduction as a dominant analytical modality for point-of-care (POC) diagnostics, replacing bulky, hospital-centered analytical instruments with portable, user-friendly devices[4].
Electrochemical sensors operate by converting chemical events at the electrode-electrolyte interface into measurable electrical signals. The approach offers several inherent advantages: (1) direct proportionality between current, potential or impedance and analyte concentration; (2) high selectivity and reversibility over broad dynamic ranges; and (3) excellent compatibility with microfabrication and wireless readout technologies[5,6]. A typical amperometric configuration consisting of a working, reference, and counter electrodes (WE, RE, CE) enables selective detection of redox species under controlled potential conditions[7]. This modular configuration is readily miniaturized into implantable or wearable formats without sacrificing analytical sensitivity[8].
The scalability and structural simplicity of electrochemical sensing platforms enable their expansion beyond metabolite and therapeutic monitoring to industrial and clinical applications. Specifically, applying electrochemical sensors in POC systems for cancer biomarkers and environmental pollutants demonstrates the versatility of this technology[3,9,10].
Extending the glucose monitoring paradigm, contemporary research focuses on non-glucose metabolic and hormonal biomarkers[11,12]. Each analyte carries distinct physiological relevance: lactate as a marker of tissue hypoxia, creatinine for renal clearance, urea for nitrogen metabolism, and cortisol as an endocrine stress hormone[13,14]. However, the physicochemical diversity of these molecules, ranging from small polar metabolites to hydrophobic steroids, requires diverse sensing strategies[15,16]. Enzymatic oxidation remains effective for redox-active metabolites. However, targets lacking intrinsic redox activity require affinity based approaches, such as those utilizing aptamers, antibodies, ionophores, or molecularly imprinted polymers (MIPs)[17-19]. The convergence of these molecular recognition strategies within electrochemical platforms defines an emerging frontier in biosensing[15].
In parallel with advances in transduction chemistry, sampling interfaces have evolved from venous or capillary blood collection toward minimally invasive and wearable platforms[20-22]. Blood remains the clinical gold standard for biomarker quantification, reflecting systemic concentrations with high accuracy[22]. Accordingly, microsampled blood, obtained via microneedle (MN) or capillary extraction, provides a practical pathway for painless and precise analysis[23,24]. In parallel, the integration of MNs, flexible substrates, and soft microelectronics has enabled continuous, extraction-free electrochemical monitoring of interstitial fluid (ISF) beneath the skin[21,25,26]. This transition from discrete to continuous sampling has reshaped design constraints: sensors must maintain electrochemical stability within complex biological matrices and ensure biocompatibility over extended wear[27,28].
This review provides a unified perspective on the electrochemical monitoring of physiologically relevant biochemicals, emphasizing how biological sampling constraints and molecular transduction strategies shape sensing performance. It also highlights how tissue architecture and ISF-blood transport kinetics influence the accessibility of analytes. Subsequent sections highlight recent device implementations that enable stable quantification of metabolites, hormones, and therapeutics. By clarifying how molecular identity governs electrochemical recognition and transduction, this review provides design principles for tailoring biosensors to diverse classes of biochemical targets. In parallel, we highlight the transition toward hybrid, multi-analyte, and closed-loop sensing systems that aim to translate continuous biochemical profiles into clinically actionable precision healthcare.
PHYSIOLOGICAL BASIS OF BIOFLUID ACCESS
Human biofluid for biosensing
Table 1 compares electrochemical biosensing platforms according to specimen type, including blood, ISF, and sweat. Blood provides the most direct reflection of analyte concentrations with a rapid response time
Comparison of blood, ISF, and sweat as biofluids for biosensing
| Biofluid | Sampling method | Mechanism | Detection targets | Reaction time | Collection difficulty | Long-term stability |
| Blood[1,30] | Invasive | Enzyme; Antibody | Glucose; Lactate; Insulin; Vancomycin | ~1 min | Moderate | Single use |
| ISF[25,30-34] | Invasive | Enzyme; Non-enzymatic; Aptamer | Glucose; Lactate; Insulin; Cortisol; Levodopa; Vancomycin | 5-15 min | Low | 7-14 days |
| Sweat[35,36] | Non-invasive | MIP; Aptamer | Glucose; Lactate; Cortisol; Vancomycin | > 5 min | Very low | 20 min to 30 days |
Skin architecture and analyte transport
Continuously tracking biomolecular dynamics within the body requires stable and physiologically representative access to biofluids. The layered architecture of human skin defines the pathways through which analytes diffuse from the bloodstream into the ISF, establishing both the opportunities and the inherent limits of ISF-based sensing[37,38]. Although small and moderately sized molecules equilibrate rapidly enough to support continuous monitoring, many large proteins and slowly transported biomarkers remain poorly represented in the ISF and therefore require direct blood-based quantification[39,40].
Human skin provides multiple access points for sensing, each located at a distinct depth and offering different trade-offs in invasiveness, analyte richness, and operational stability. Structurally, the skin comprises three principal layers-the epidermis (0.1-0.3 mm), dermis (0.6-3 mm), and subcutaneous tissue (2-5 mm) [Figure 1A]. The dermis and subcutis contain a dense capillary network with 50-100 μm inter-capillary spacing, and ISF fills 15%-20% of the extracellular space within these compartments[37,41].
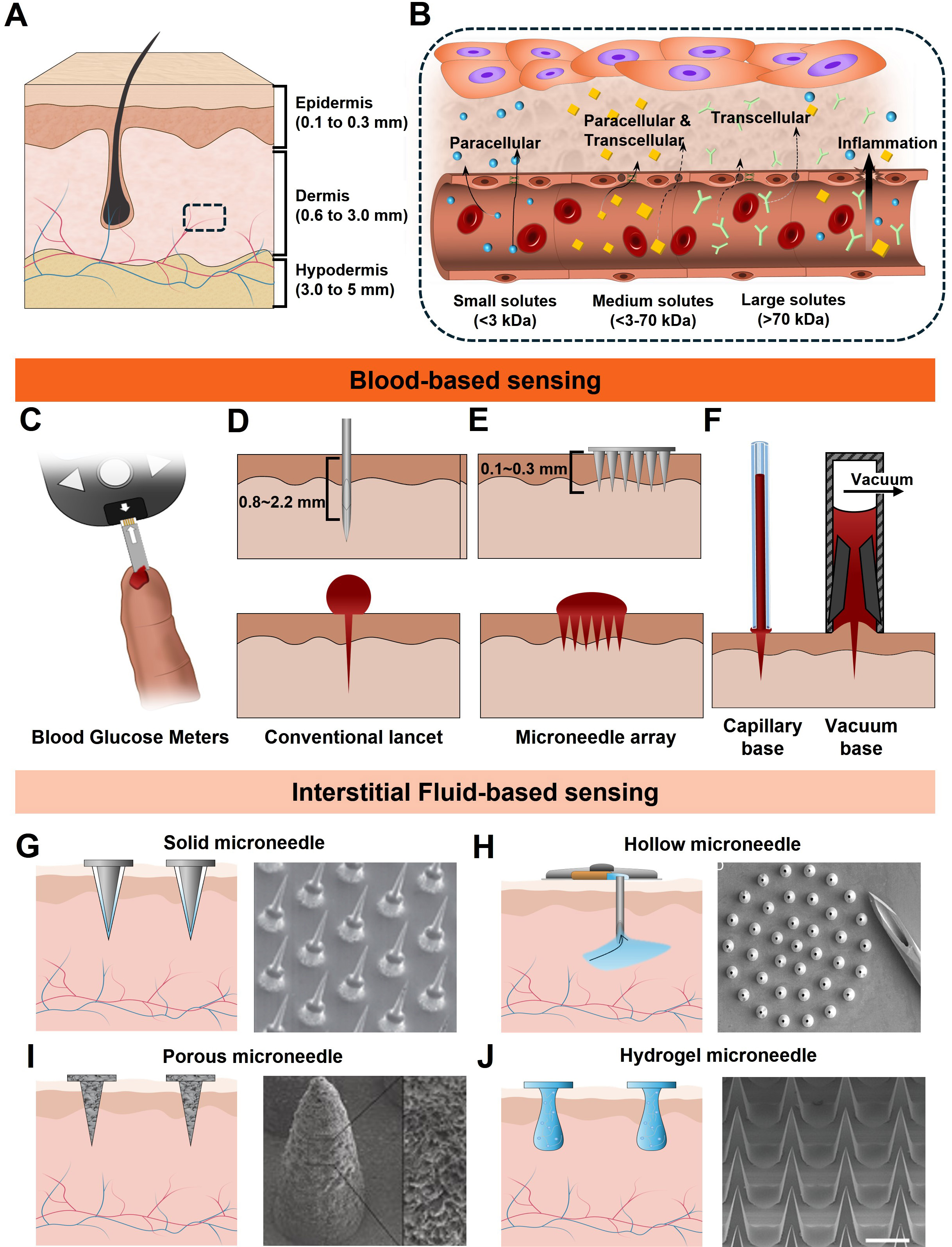
Figure 1. Biomarker sensing strategies in the human body. (A) Overall anatomical structure of the skin; (B) The capillary interface consists of a single layer of endothelial cells and inter-endothelial junctions, regulating the exchange of metabolites between blood and ISF. Sampling strategies for blood (C-F) and ISF (G-J); (C) Schematic of a commercial blood glucose meter; (D) Single-needle lancet method for blood extraction; (E) Penetration schematic of a microscale needle array; (F) Conceptual illustration of ampule based blood collection from exuded droplets; (G) Solid MNA patch for minimally invasive ISF extraction; (H) Porous and surface-functionalized MN patch; (I) Hollow MN utilizing capillary action for ISF uptake; (J) Hydrogel-based MN patch for passive ISF absorption. ISF: Interstitial fluid; MN: microneedle.
Exchange of metabolites between the bloodstream and ISF occurs predominantly via passive diffusion across the capillary endothelium [Figure 1B][42]. Two major pathways govern this transport: the paracellular and transcellular pathways[37,41,43]. In the paracellular route, analytes traverse the inter-endothelial junctions (IEJs), which permit the passage of small molecules - generally molecules smaller than ~70 kDa - through diffusion and advection[44]. The transcellular pathway accommodates both hydrophobic and hydrophilic analytes: hydrophobic molecules diffuse through the lipid bilayer, whereas hydrophilic species are transported via intracellular vesicular mechanisms[45].
Inflammation or vascular injury can disrupt endothelial integrity, leading to nonspecific leakage of blood constituents and thereby increasing the levels of blood constituents in ISF. The diffusion coefficient, molecular weight, and polarity of a given analyte largely determine its equilibration time between blood and ISF, consistent with the temporal delays observed in prior ISF-based sensing studies[32,46-51]. Consequently, most blood-borne analytes appear in the ISF through diffusion and occasional leakage, providing the fundamental basis for ISF sampling in continuous monitoring systems.
Microsampled blood access and capillary physiology
Blood analysis remains the quantitative gold standard for systemic physiological assessment[52-54]. The homogeneous distribution of circulating analytes ensures highly reproducible sampling conditions and supports traceable quantification[55]. Furthermore, the uniform concentration profile across the bloodstream minimizes spatial variability, thereby avoiding the sampling-dependent gradients that frequently complicate ISF-based sensing modalities.
In conventional clinical diagnostics, several milliliters of venous blood are typically collected to enable multiplexed biomarker analysis using centralized laboratory instrumentation[56,57]. Such procedures require trained personnel, sterile phlebotomy equipment, and dedicated infrastructure, limiting their suitability for frequent or near-patient monitoring[58]. In contrast, the small-volume sampling strategies discussed here are intended for decentralized, user-operated measurements of one or a few targeted biomarkers at the point of care. A representative example is the capillary blood testing used in commercial glucose meters [Figure 1C][59-61].
Despite the substantially reduced volume, a single ~1 μL capillary blood sample can contain biomarkers at nanomolar concentrations, enabling electrochemical sensing with a high signal-to-noise ratio (SNR). Historically, capillary blood acquisition has relied on spring-loaded mechanical lancets penetrating approximately 0.8-2.2 mm to access the superficial capillary bed [Figure 1D]. Recent iterations incorporating silicon-etched tips and optimized bevel geometries have decreased penetration force and improved depth control, resulting in significantly reduced pain[62]. Disposable lancets remain inexpensive, reliable, and widely implemented in commercial BGMs. However, they induce open bleeding and, when penetrating into the mid-to-lower dermis (0.5-2 mm), can activate dense nociceptor networks, presenting an infection risk and user discomfort[63].
To overcome these limitations, emerging MN and microstructured needle array technologies can extract comparable sample volumes at penetration depths below 100 μm, enabling access to the superficial capillary plexus while largely avoiding nociceptors [Figure 1E][64,65]. Although conventional fingertip lancets typically yield ~3.1 μL of blood, alternative anatomical collection sites often produce smaller volumes[62]. These challenges can be mitigated by sample acquisition strategies that improve collection efficiency. Examples include capillary-driven microtubes and controlled vacuum-assisted extraction, as described in Figure 1F.
ISF access and continuous monitoring
Blood-based electrochemical platforms enable the detection of a broad spectrum of analytes, ranging from small metabolites such as glucose and lactate to larger biomolecules including peptides and proteins. Appropriate surface functionalization further expands this detection capability across diverse molecular targets[66-69]. Despite this versatility, long-term monitoring requires substantial material and clinical resources, and real-time feedback is often limited. Repeated sampling also poses a risk of iatrogenic anemia in individuals with low blood volume, including neonates and patients with preexisting anemia. These limitations have driven recent research toward continuous monitoring and closed-loop feedback systems, supported by rapid advances in biointegrated sensor technologies.
ISF represents a highly suitable biofluid for continuous monitoring applications. Its ionic strength (~150 mM Na+), physiological pH (~7.4), and overall small-molecule biomarker composition closely resemble those of plasma, while avoiding the clot formation that complicates conventional blood sampling[33,41,43]. CGMs exemplify ISF-based sensing systems, typically operating with measurement intervals of 1-5 min and exhibiting a physiological lag of approximately 5-15 min relative to blood glucose levels[33,70,71]. Owing to its minimally invasive accessibility and capacity for long-term autonomous operation without repeated blood withdrawal, ISF analysis holds significant promise for future clinical diagnostics and chronic disease management.
The geometry, material composition, and penetration depth of a microneedle array (MNA) critically determine its ability to reliably access ISF, directly influencing signal quality, mechanical stability, and biocompatibility[64,72]. Penetration depths shallower than ~0.5 mm often result in impedance drift due to local dehydration, whereas depths exceeding ~6 mm can induce inflammatory encapsulation and alter diffusion profiles[34]. Commercial CGMs, such as the Dexcom G7 and Freestyle Libre 3, position the WE within the deep dermal compartment, approximately 5 mm below the skin surface. This configuration helps ensure consistent mechanical-electrical coupling with the ISF. These optimized configurations minimize bleeding and nociceptive activation, enabling pain-free long-term wear while maintaining high analytical reliability through continuous ISF replenishment[73].
Microneedles function as minimally invasive channels that interface the ISF with the sensing element. Their geometric design governs ISF accessibility, operational lifetime, and the achievable SNR. Among available microneedle configurations, solid MNs represent the simplest configuration and may serve either as direct transdermal electrodes or as perforation tools to enhance ISF transport [Figure 1G]. Their strong mechanical integrity and compatibility with metal or metal-oxide functional coatings make them well suited for long-term autonomous sensing in wearable formats[74,75]. Commercial CGM systems use a related design. In these devices, a flexible electrochemical WE is inserted into the dermis through a temporary stainless steel guide needle, which is retracted after proper placement. The implanted sensing element then maintains stable electrical contact with the ISF, enabling continuous electrochemical measurements over multiple days without repeated insertion.
Various MN platforms enabling intermittent ISF sampling have been proposed for distinct analytical and clinical applications. Hollow MNs feature a central microscale channel within the needle structure [Figure 1H]. This hollow channel allows ISF extraction during application and enables precise drug delivery[76]. Their ability to transport larger biomolecules and therapeutics makes them advantageous for transdermal administration. However, challenges such as stratum corneum intrusion, lumen blockage, and limited mechanical strength remain significant barriers to clinical translation.
Porous MNs [Figure 1I] are typically fabricated from polymers or metal-inorganic composites and contain interconnected micropores throughout their structure. Their high surface-area-to-volume ratio facilitates enhanced sensitivity via surface functionalization, enabling high-performance biochemical sensing[77,78]. When integrated with iontophoresis, porous MNs can actively extract ISF into a sensing chamber, improving sampling efficiency.
Hydrogel MNs are produced by molding hydrogel precursors followed by freeze-drying or conventional drying processes to form a porous conical structure [Figure 1J]. The stiffness of the MNs can be tuned by adjusting polymer crosslinking density. In the dry state, hydrogel MNs exhibit sufficient rigidity for skin penetration. Once inserted, they rapidly swell upon absorbing ISF, becoming softer while simultaneously extracting significant fluid volumes[79,80]. Solution based fabrication methods readily accommodate the incorporation of functional materials, positioning hydrogel MNs as a promising platform for next-generation wearable and patch based biosensing systems. As a case in point, GhavamiNejad et al. demonstrated that hydrogel-based MN platforms provide a mechanically compliant interface for transdermal sensing[81]. The soft and biocompatible hydrogel structure enables painless insertion and efficient extraction of ISF, while maintaining intimate contact with the surrounding tissue. Significantly, the osmotic swelling of the hydrogel enables continuous ISF absorption, thereby lowering the charge transfer resistance and increasing the interfacial capacitance. Furthermore, the flexible hydrogel MNA maintains stable electrochemical signals under repeated mechanical deformation, demonstrating its suitability for reliable real-time monitoring in dynamic biological environments.
Blood and ISF constitute two physiologically interconnected yet operationally distinct media for biosensing. Blood analysis provides unrivaled analytical breadth and accuracy; however, its dependence on invasive access and intermittent collection constrains its suitability for real-time and long-term monitoring. By contrast, ISF can be accessed at shallow depths in the dermis using minimally invasive MN-based interfaces, enabling continuous electrochemical measurement without repeated phlebotomy or clinical assistance. Nevertheless, ISF primarily supports the detection of small and moderately sized molecules. Larger proteins and slowly transported biomarkers do not equilibrate reliably and therefore remain more accurately quantified in blood. Unlocking the full capability of ISF-based monitoring demands precise control over the biophysical environment surrounding the electrode. Solute diffusion kinetics, tissue hydraulic permeability, and depth-dependent electrical impedance collectively determine signal stability, noise susceptibility, and calibration fidelity. Accordingly, the choice between blood- and ISF-based sensing should be guided by the intended analytical scope, required temporal resolution, and user burden.
BIOMARKERS ACCESSIBLE VIA TRANSDERMAL BIOFLUID SENSING
Human physiology maintains homeostasis through tightly regulated biochemical networks that couple metabolic activity across multiple organs. Because blood and ISF are in continuous exchange through capillary diffusion, clinically relevant biochemical information can, in principle, be obtained from either medium. ISF provides a minimally invasive alternative for the long-term monitoring of molecules that achieve reliable equilibrium with blood. Conversely, biomarkers with slow transport kinetics are more accurately quantified through direct blood analysis. Biomarkers of current interest that can be measured through microsampled blood or ISF can be broadly grouped into two categories. These include (1) endogenous metabolites that reflect physiological status and (2) exogenous or pharmacological molecules whose concentrations determine therapeutic efficacy and safety. This section summarizes key molecular targets currently accessible through electrochemical biosensing and highlights their relevance in clinical decision-making.
The clinical utility of electrochemical biosensors depends on their ability to accurately discriminate between physiological concentration ranges in healthy individuals and pathological ranges associated with disease states [Table 2]. For example, glucose concentrations in healthy adults are typically 3.2-9.2 mM in ISF, whereas patients with diabetes exhibit a broader range of 1.99-22.2 mM[6]. Successful clinical application necessitates high sensor stability for long-term wear (e.g., 14 days) and algorithmic compensation for physiological time lags between blood and ISF. Similarly, normal blood lactate concentrations fall within 0.5-1.5 mM, whereas levels exceeding 2.0 mM indicate sepsis, shock, or systemic hypoperfusion[6,82,83]. This makes real-time lactate monitoring critical for acute care. Translating lactate detection to sweat further requires the mitigation of signal interference caused by varying sweat rates to maintain user compliance.
Physiological reference range of metabolites in biofluids (units: mM)
| Biomarker (disease) | Biofluid | Physiological range (healthy) | Pathological range (abnormal) | Standard lab method (gold standard) |
| Glucose (Diabetes) [6] | Blood | 3.9-5.6 | > 7.0 | Amperometry |
| ISF | 3.2-9.2 | 2.0-22.2 | - | |
| Sweat | 0.06-0.11 | 0.01-1.0 | - | |
| Lactate (Sepsis/Shock/COPD) [6,82,83] | Blood | 0.5-1.5 | > 2.0 | Blood gas analyzer |
| ISF | 0.5-1.5 | > 3.4 | - | |
| Sweat | 16-30 | 0.01-1.0 | - | |
| Urea (CKD, ESRD) [86,87] | Blood | 5.37-7.71 | 18.75-26.25 | Enzymatic |
| ISF | 3.9-4.9 | 20.69-23.8 | - | |
| Sweat | 22.2 | 22.02-33.25 | - | |
| Uric acid (Gout, CKD, Hyperuricemia) [6,84,85] | Blood | 0.2-0.42 | > 0.42 | HPLC/Colorimetry |
| ISF | ~0.36 | > 0.36 | - | |
| Sweat | ~0.02 | 0.018-0.036 | - | |
| Creatinine (CKD) [86,88,89] | Blood | 0.045-0.11 | > 0.2 | Jaffé Reaction/Enzymatic |
| ISF | 0.045-0.11 | > 0.2 | - | |
| Sweat | 0.01-0.018 | 0.06-0.2 | - |
Uric acid also exhibits disease-dependent variation; blood concentrations increase substantially in patients with hyperuricemia, from normal levels of 0.2 to 0.42 mM[6,84,85]. Reliable blood monitoring necessitates long-term baseline drift management, while sweat-based sensors present a promising non-invasive alternative to frequent blood draws. Furthermore, comprehensive renal function evaluation requires precise quantification of urea and creatinine, often utilizing the blood urea-to-creatinine ratio. Healthy blood urea levels (5.37-7.71 mM) increase significantly in end-stage renal disease to 18.75-26.25 mM[86,87]. Creatinine similarly increases from normal levels (0.045-0.11 mM) to > 0.2 mM during renal failure, demanding high target selectivity[86,88,89].
Given the close correlation between systemic blood and ISF concentrations, continuous ISF monitoring has emerged as a suitable approach for wearable kidney monitoring systems. Conversely, sweat-based measurement involves significant analytical challenges, notably concentration variability dependent on sweat rates. For instance, the extremely low physiological concentration of sweat creatinine (0.01-0.018 mM) explicitly demands ultra-high sensitivity and selectivity to detect pathological elevations (0.06-0.2 mM)[88]. Consequently, the analytical specifications of biosensors - sensitivity, dynamic range, temporal resolution, and stability - must be rigorously tailored to specific biomarkers and their respective pathological windows. This is essential for reliably capturing both normal physiological fluctuations and disease progression.
Metabolites
Energy metabolism in humans arises from a coordinated network linking the liver, kidneys, skeletal muscle, and other organs [Figure 2A]. These tissues exchange intermediates through core biochemical circuits, including glucose homeostasis, the Cori cycle, nitrogen metabolism, and the creatine-creatinine pathway. Metabolites generated through these pathways serve as clinically informative biomarkers.
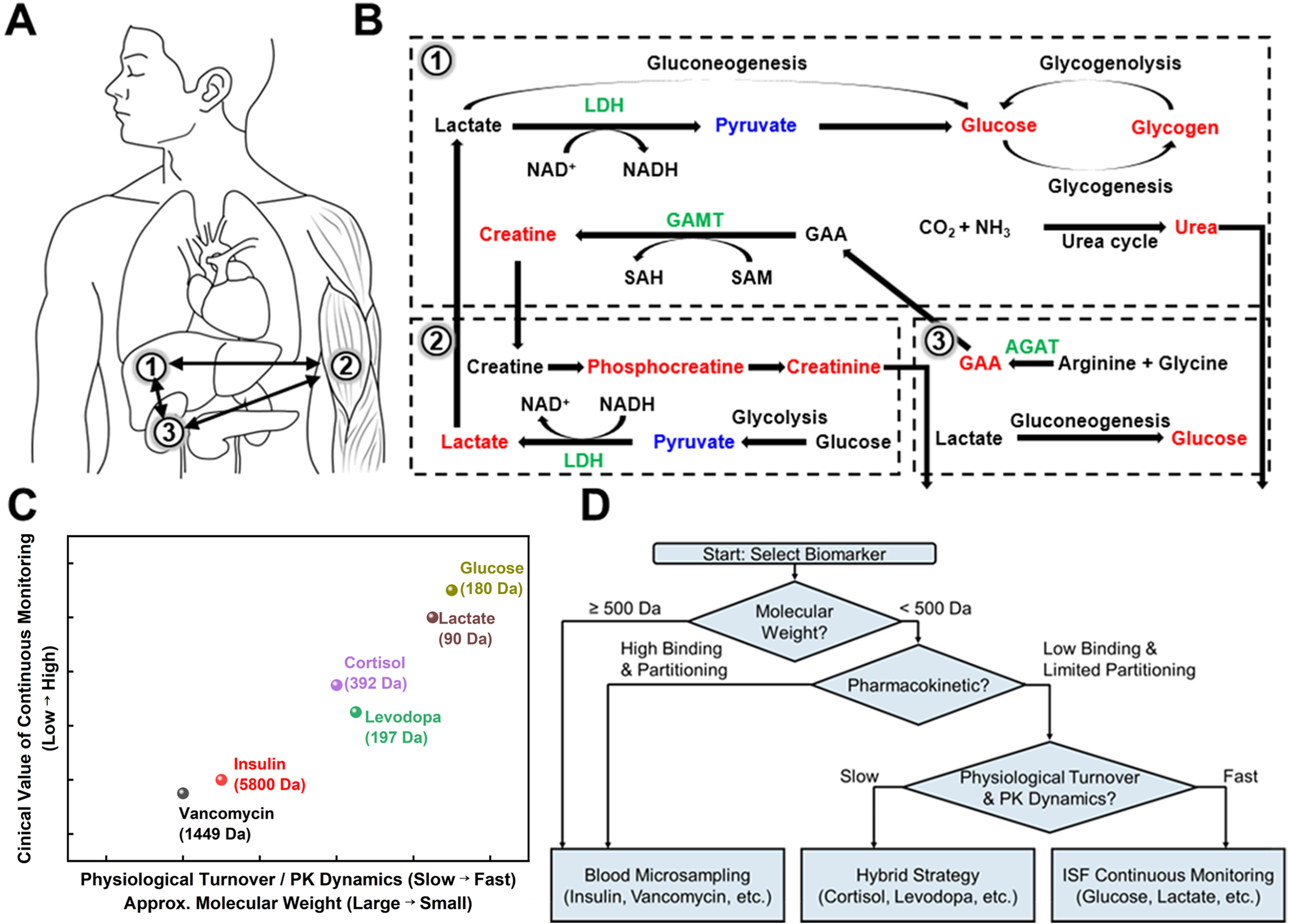
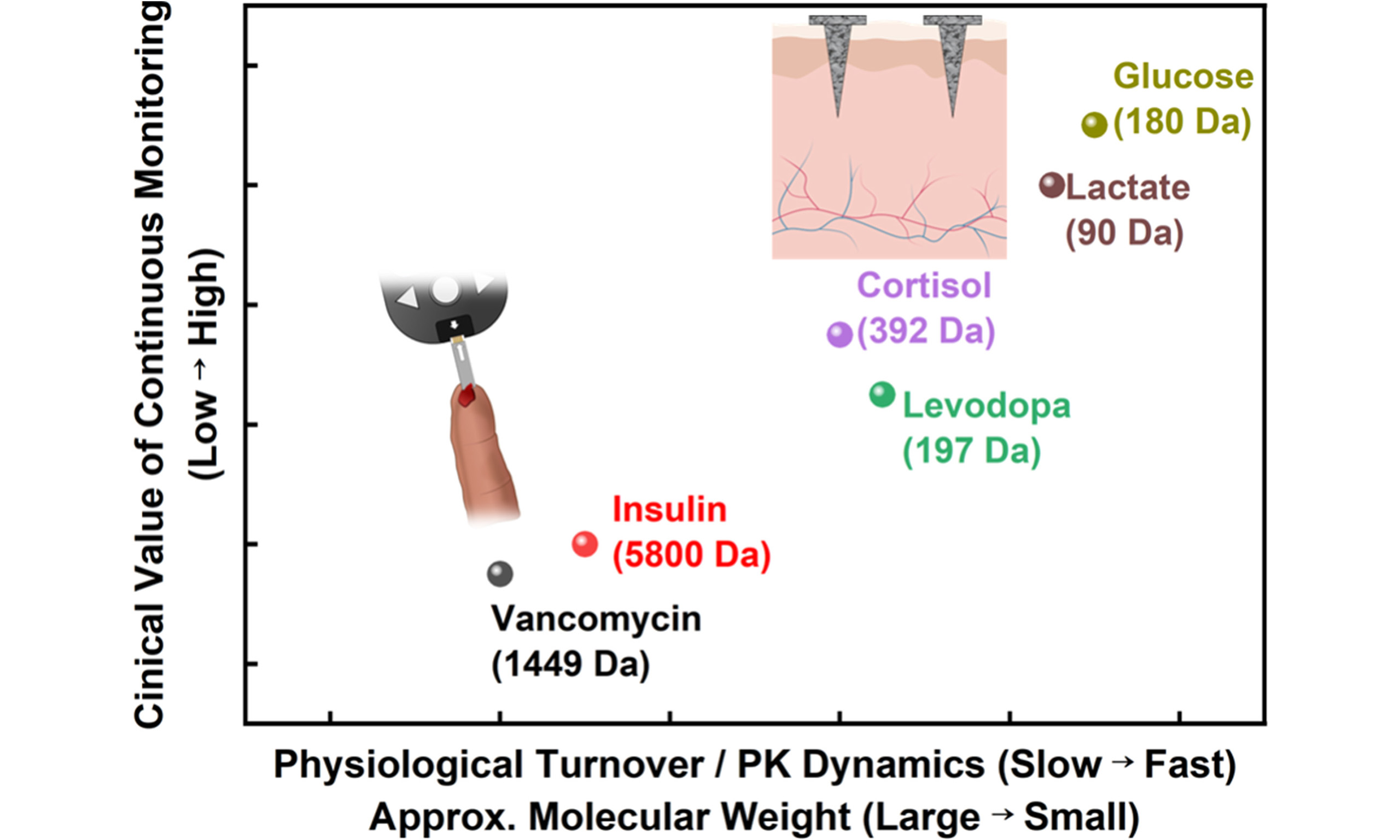
Figure 2. Biological metabolic pathways and sensing strategy selection. (A and B) Metabolic processes in the human body; (A) Schematic illustration of major target organs associated with key biomarkers: (1) liver, (2) muscle, and (3) kidney; (B) Metabolic flow map of glucose, creatine, and lactate. These pathways illustrate the dynamic interactions between organ-specific metabolism and systemic physiological environments; (C) Relationship between biomarker molecular mass, PK dynamics, physiological turnover, and the resulting need for continuous monitoring; (D) Decision-making framework for selecting optimal sensing strategies based on the physicochemical and physiological profiles of target biomarkers. PK: Pharmacokinetic; NAD+: nicotinamide adenine dinucleotide (oxidized form); NADH: nicotinamide adenine dinucleotide (reduced form); LDH: lactate dehydrogenase; SAH: S-adenosyl-L-homocysteine; SAM: S-adenosyl-L-methionine; GAA: guanidinoacetate; GAMT: guanidinoacetate N-methyltransferase; AGAT: arginine: glycine amidino transferase.
The liver orchestrates systemic glucose regulation by balancing glycogenesis, glycogenolysis, and gluconeogenesis [Figure 2B]. Postprandial insulin promotes hepatic glucose uptake and glycogen storage, whereas fasting or stress triggers glucagon and epinephrine-mediated glycogenolysis[90]. During prolonged fasting or anaerobic exercise, the liver converts lactate, alanine, and glycerol into glucose via gluconeogenesis. In particular, lactate is recycled into glucose through lactate dehydrogenase- and pyruvate carboxylase-dependent steps in the Cori cycle[91]. Liver dysfunction disrupts these pathways, leading to impaired lactate clearance. Consequently, elevated lactate is a hallmark of sepsis, hypoxia, and hepatic injury.
Creatine metabolism exemplifies interorgan coordination[92]. The kidney synthesizes guanidinoacetate (GAA) via arginine: glycine amidino transferase (AGAT), and the liver methylates GAA to creatine through guanidinoacetate N-methyltransferase (GAMT). Skeletal muscle then phosphorylates creatine to phosphocreatine (PCr) for rapid adenosine triphosphate (ATP) regeneration. Creatine and PCr spontaneously cyclize to creatinine, which is filtered by the kidney. Reduced renal function decreases creatinine clearance and elevates circulating creatinine, a central biomarker of kidney dysfunction.
Nitrogen detoxification further integrates hepatic and renal functions. The liver converts ammonia to urea via the urea cycle, and the kidney subsequently excretes urea[93]. Liver failure leads to hyperammonemia and impaired urea production, whereas kidney failure results in reduced urea excretion and elevated blood urea.
Together, these organ systems regulate glucose balance, energy buffering, and nitrogen disposal, producing metabolites that sensitively reflect hepatic and renal status. Because these small molecules rapidly equilibrate between blood and ISF, they constitute robust targets for continuous wearable monitoring. This enables real-time assessment of perfusion, metabolic activity, and organ function. Additionally, investigating the complex correlations between multiple biomarkers will be essential for enhancing diagnostic specificity in multi-organ pathologies[94]. A representative example is the metabolic interdependence among glucose, lactate, and creatinine.
Hormones and other endogenous signaling molecules
Hormones constitute a distinct class of endogenous biomarkers whose circulating concentrations encode endocrine regulation, physiological stress, and metabolic homeostasis. The suitability of hormones for transdermal sensing is heavily dictated by their molecular size, hydrophobicity, and carrier-protein binding affinity. These physicochemical properties govern their transport across the capillary endothelium and subsequent partition into the ISF. Some steroid and amine hormones are accessible in ISF and compatible with minimally invasive sensing, whereas many peptide hormones require direct blood sampling for accurate quantification.
For example, cortisol, a hydrophobic glucocorticoid synthesized in the adrenal cortex, diffuses across cell membranes and equilibrates sufficiently between blood and ISF to enable minimally invasive sensing[95-97]. Transdermal cortisol measurements have been demonstrated using iontophoretic extraction, MN-assisted sampling, and electrochemical platforms employing affinity receptors or MIPs. Cortisol reflects hypothalamic-pituitary-adrenal (HPA) axis activity and exhibits rapid diurnal dynamics. Accordingly, real-time monitoring offers a window into physiological and psychosocial stress states, as well as adrenal dysfunction[98].
In contrast, insulin (Mw ≈ 5.8 kDa) exhibits limited passive transport into ISF due to its size and dependence on receptor-mediated clearance pathways[37]. Although insulin can be detected using MN-based sampling or microdialysis techniques, its ISF concentrations lag substantially behind plasma levels and often fall below the detection limits of current electrochemical platforms[99,100]. As a result, insulin remains more accurately quantified through microsampled capillary blood, particularly for applications requiring precise pharmacokinetic profiling or closed-loop glycemic control[101].
Pharmacological and other exogenous molecules
Beyond endogenous biomolecules, several exogenous or therapeutic molecules present in circulation can also serve as informative biosensing targets. Depending on their physicochemical properties, these molecules may be detected using either microsampled blood or ISF. Small and moderately sized drugs that exhibit rapid capillary-tissue exchange, such as levodopa used in Parkinson’s disease management, readily diffuse into ISF[102]. Other clinically important agents, however, display limited ISF penetration due to their molecular size, hydrophobicity, or strong protein binding. For example, vancomycin (Mw ≈ 1.45 kDa) is a narrow-therapeutic-window antibiotic that requires precise dosage adjustments. Owing to its restricted diffusion across the endothelial barrier, it is most accurately quantified using microsampled capillary blood rather than ISF. Similar constraints apply to many chemotherapeutics, peptide hormones, and biologics. Their tissue distribution and slow equilibration kinetics often preclude ISF-based measurement. The suitability of each compound for ISF versus blood-based sensing is thus determined largely by its molecular size, hydrophobicity, and binding characteristics. Detailed electrochemical sensing strategies for these pharmacological targets, following the discussion of metabolite sensing, are provided in subsequent sections.
Considerations for selecting blood versus ISF sampling
Beyond molecular accessibility, the choice between blood and ISF sampling is primarily dictated by the clinical informativeness of temporal resolution and the quantitative fidelity required for decision-making. Continuous ISF monitoring is most valuable when rapid fluctuations in biomarker levels directly inform physiological control or therapeutic adjustment. Glucose and lactate are representative examples[103,104]. In such cases, high frequency data capture enables the detection of transient excursions that would be missed by intermittent testing. Conversely, for diagnostics based on absolute thresholds (e.g., renal function or therapeutic drug monitoring), intermittent microsampled blood analysis is sufficient and frequently provides greater diagnostic clarity. Thus, the sampling strategy is fundamentally shaped by the clinical context in which the biomarker is applied.
A second key determinant is the physiological turnover and pharmacokinetic (PK) dynamics of the target molecule. Together, these factors define the characteristic time scale of production, systemic distribution, and clearance. Biomarkers with rapid turnover benefit most from continuous monitoring, as their concentrations vary on the time scale of minutes to hours and carry immediate physiological significance. In contrast, slowly varying hormones and drugs with long elimination half-lives exhibit limited incremental benefit from minute-scale ISF tracking, even when technically detectable. For many pharmacological agents, accurate dosing decisions depend on resolving systemic PK parameters rather than local ISF fluctuations. Accordingly, microsampled blood remains the preferred analytical matrix for high-precision therapeutic guidance. Molecular weight serves as a primary, though not exclusive, determinant of these dynamics, with additional contributions from protein binding, tissue partitioning, and metabolic clearance.
These relationships are summarized in Figure 2C. The figure presents a decision map for selecting blood versus ISF sampling based on physiological turnover, which is correlated with PK dynamics and molecular properties, as well as the clinical value of continuous monitoring. High turnover metabolites such as glucose and lactate occupy the ISF continuous domain, whereas slowly varying or macromolecular targets such as insulin and vancomycin are confined to the blood-based regime[21,37,43]. Intermediate targets, including cortisol and levodopa, occupy a hybrid region in which ISF access is feasible, but continuous monitoring provides only limited additional clinical benefit. Importantly, quantitative robustness and clinical standardization further constrain these choices. ISF-based sensing must contend with lower analyte concentrations, depth-dependent variability, biofouling, and long-term signal drift. By contrast, blood microsampling aligns more directly with established clinical reference ranges and regulatory validation frameworks. Accordingly, the optimal strategy should be regarded as a biomarker-specific, application-driven selection. Hybrid monitoring paradigms that integrate continuous ISF sensing with periodic blood microsampling represent the most reliable route toward clinically interpretable electrochemical biosensing. Figure 2D summarizes the decision framework for selecting blood versus ISF sampling across diverse biomarker classes.
ELECTROCHEMICAL SENSING
Disease diagnosis based on biomarkers requires both real-time monitoring and precise quantitative analysis. In real-time biomonitoring environments, sensing systems must not only provide high analytical performance but also satisfy the requirements of miniaturization and wearability. Mass spectrometry offers exceptional sensitivity and molecular specificity; however, its application in wearable diagnostics remains limited[105]. Typically, ISF must be extracted using hydrogels or MN and then transported to benchtop instrumentation for analysis. This multi-step process prevents its use in continuous, on-site monitoring systems.
Optical sensing methods including surface-enhanced Raman spectroscopy (SERS), colorimetric assays, and fluorescence based detection face practical limitations in wearable applications [Figure 3A][106,107]. For instance, a previous study demonstrated a SERS platform based on a functionalized silver-coated PMMA MNA that achieved approximately 93% diagnostic reliability in a diabetic mouse model[107]. However, the system required complex external instrumentation (laser source, Raman spectroscopy equipment, and a video microscope). This requirement restricts the utility of SERS for routine or portable applications. Similarly, a multi-analyte MN colorimetric system reported by Zhu et al., composed of cross-linked methylhydroxyapatite (MeHA) and soluble osmolytes, enabled simultaneous detection of pH, glucose, lactate, and cholesterol[108]. However, signal interpretation relies on color changes observed by the naked eye or a camera. This dependency makes the method highly susceptible to variations in ambient lighting and results in a lack of precision for accurate quantitative measurements[107-111]. In real-time monitoring applications, such reliance on complex external instrumentation and limited quantitative resolution increases device size and operational complexity. These factors potentially cause user discomfort and restrict sensitivity in trace metabolite detection.
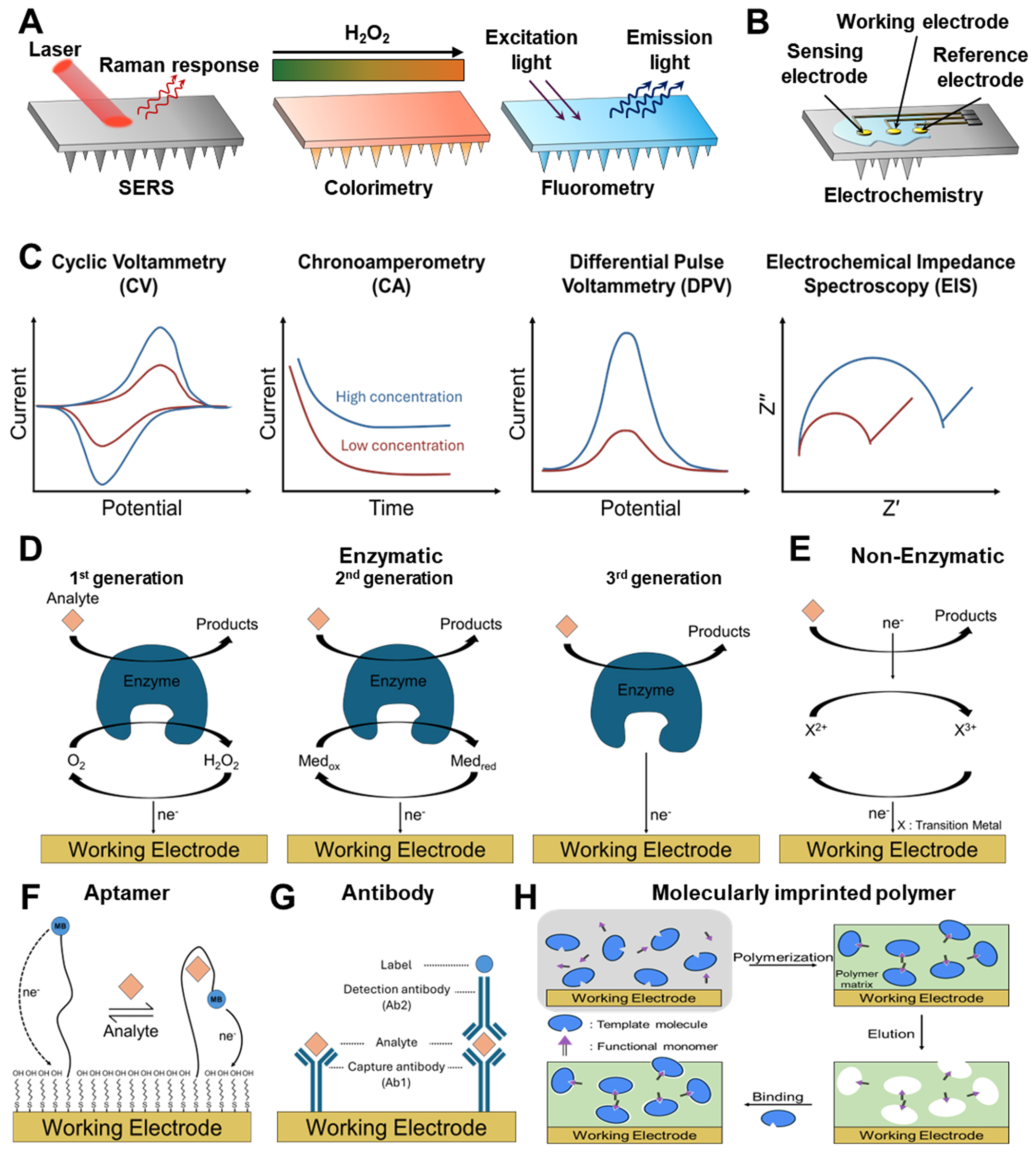
Figure 3. Principles and techniques of electrochemical biomarker sensing. (A and B) ISF analysis method based on optical (A) and electrochemical reaction (B); (C) Overview of electrochemical biosensing measurement techniques, including CV, CA, DPV, and EIS, along with their characteristic electrical signal outputs; (D-H) Various sensing mechanisms: (D) enzyme-based electrochemical biosensors across different generations, (E) redox-active and non-enzymatic catalytic sensors, (F) aptamer-based sensors, (G) antibody-based immunosensors, and (H) MIP-based biosensors. ISF: Interstitial fluid; MIP: molecularly imprinted polymer; SERS: surface-enhanced Raman spectroscopy.
In contrast, electrochemical sensing directly converts biochemical interactions into electrical signals, enabling rapid and precise quantitative analysis without requiring bulky signal-conversion equipment. Moreover, electrochemical sensors operate with minimal power consumption by detecting small variations in current or potential. This efficiency makes them well-suited for long-term wearable monitoring without significant heat generation. Accordingly, this section introduces the fundamental principles of electrochemical analysis and highlights their implementation in practical wearable biosensing platforms.
Signal transduction modalities
Early ISF sensing studies typically relied on extracting ISF through MN and subsequently transferring the collected sample to an external test tube or detection chamber for analysis. This workflow inherently required an additional transport step, delaying detection and complicating integration with real-time feedback systems. Electrochemical transduction is the most widely adopted quantitative method for direct on-site signal transduction. Electrochemical biosensors convert charge transfer phenomena occurring at the electrode-electrolyte interface into electrical signals such as current, potential, or impedance. Even when the analyte does not directly perform oxidation or reduction reactions, its binding to the electrode surface can alter properties such as the electron transfer rate, interface structure, ion mobility, and charge accumulation. These changes can then be electrically quantified.
Electrochemical sensors generally employ either two-terminal or three-terminal configurations, depending on the mode of operation-amperometric, potentiometric, or impedimetric. Two-terminal systems integrate the WE and the CE/RE into only two electrodes. This architecture simplifies circuit design and is advantageous for compact, wearable implementations. However, because the CE simultaneously serves as the RE, the applied potential and measured current are not independently controlled. This lack of independent control limits the precision of potential regulation. In contrast, three-terminal systems include WE, RE, and CE, and are optimized for precise potential control and quantitative accuracy [Figure 3B]. The RE (commonly Ag/AgCl) maintains a highly stable potential, providing a constant reference against which the WE potential is controlled. The CE supplies or withdraws electrons necessary to sustain redox reactions at the WE and is fabricated from chemically inert materials such as Pt, Au, or stainless steel[112-115]. The WE serves as the site of direct electrochemical reaction, where enzymes, conductive nanomaterials, redox mediators, or catalytic materials are immobilized. Within this configuration, redox events, electron transfer, and ion transport occur at the WE-biofluid interface. These phenomena enable immediate quantification of the target biomarker through changes in current, potential, or impedance.
Figure 3C shows the representative waveforms and concentration-dependent responses of major electrochemical measurement techniques, including cyclic voltammetry (CV), chronoamperometry (CA), differential pulse voltammetry (DPV), and electrochemical impedance spectroscopy (EIS). CV involves sweeping the potential of the WE at a fixed rate while measuring the current response. This provides an intuitive understanding of key electrochemical properties of the analyte, such as its oxidation/reduction potential, electron-transfer reversibility, and interfacial catalytic activity[116]. It is the standard method for understanding the fundamental mechanisms of electrochemical reactions[117]. CA applies a fixed potential and monitors the decay of current over time. This method is useful for tracking real-time concentration changes in enzyme-based sensors or H2O2-based reactions[118].
DPV is a high-sensitivity quantitative technique that superimposes small pulses onto a staircase potential, selectively extracting and amplifying Faradaic currents[119]. It significantly suppresses background currents, making it well suited for low-concentration analysis of hormones, drugs, and metabolites. Accordingly, it is one of the most widely used electrochemical platforms for the quantitative analysis of small molecules and proteins[120,121]. EIS involves measuring impedance over a wide frequency range to quantify charge transfer resistance, double-layer capacitance, and interfacial reaction resistance[122]. Because changes in interfacial structure induced by target binding are directly reflected in impedance, EIS is a core readout technique in label-free sensors such as aptamer-, antibody-, and MIP-based biosensors[123].
CV, CA, DPV, and EIS each offer distinct strengths and are widely used as essential measurement tools in electrochemical signal transduction. They are applied across a range of sensor platforms, including enzyme-based, aptamer-based, antibody-based, non-enzymatic (redox-active), and MIP-based sensors. Together, these four techniques play complementary roles in quantifying changes in electron-transfer behavior at the electrode interface.
Molecular recognition mechanisms
Table 3 summarizes the quantitative performance of representative electrochemical biosensors and their suitable target biomarkers according to sensing mechanism. Traditionally, enzyme-based biosensors have dominated the field of electrochemical biosensing because of their rapid response and high selectivity. Owing to their efficient signal transduction, enzyme-based platforms typically exhibit response times within a few minutes and detection limits as low as 80 nM for glucose[1,124-127]. These characteristics highlight their suitability for real-time monitoring. However, enzyme degradation and oxygen dependence remain major limitations for long-term operation, reducing their utility in continuous trend monitoring. Antibody-based biosensors can achieve exceptionally low detection limits, down to 0.024 pg/mL, while maintaining excellent selectivity[128]. Nevertheless, their irreversible binding characteristics and limited operational lifetime make them less suitable for continuous monitoring and more appropriate for single-use, high-precision diagnostic applications[129].
Comparison of characteristics by mechanism used in biosensors
| Sensing mechanism | Analytes | Response time | Detection range |
| Enzyme[1,124-127] | Glucose; Lactate; Creatinine; Urea | 30-90 s | > 80 nM 0.25-35 mM 0.01-12 μM 1-16 mM |
| Antibody[128] | Insulin | 15-30 min | > 0.024 pg/mL |
| Aptamer[25,36,130,131] | Cortisol; Insulin; Vancomycin; Tobramycin | 10 s | 1-1,000 nM 0.01-4 nM 1-50 μM 2-100 μM |
| Non-enzymatic[114,132] | Glucose; Levodopa | ~82 s | > 20 mM 5-300 μM |
| MIP[35,133,134] | Cortisol; Vancomycin | 8-15 min | > 181 pM > 10 pM |
Recent advances in electrode architecture, particularly the incorporation of three-dimensional microstructures and nanomaterials, have substantially improved the SNR of aptamer-based biosensors[25,36,130,131]. For example, a recent study reported insulin detection over a range of 0.01-4 nM within approximately 10 s[25]. The reversible binding behavior of aptamers also highlights their potential for long-term, repeatable monitoring. Similar progress in materials and interface engineering has also revitalized electrochemical sensing platforms that were previously limited by poor sensitivity or slow response. Non-enzymatic redox sensors were once regarded as secondary options because of insufficient selectivity and sluggish kinetics. However, advances in porous supports and catalytic surface design have now enabled glucose sensitivities as high as 22.99 μA/mM[132].
A comparable trend is observed in molecularly imprinted polymer (MIP)-based sensors[35,133,134]. Although these systems have traditionally suffered from slow mass transport and limited temporal resolution, recent studies have shown that the introduction of three-dimensional porous architectures can markedly improve their performance. For example, such an approach enabled vancomycin detection with a limit of detection as low as 10 pM[134]. These results underscore the promise of MIP-based sensors for long-term, high-sensitivity monitoring in biofluids such as sweat, saliva, and tears. This potential is further supported by the intrinsic chemical stability of inorganic and polymeric platforms.
Collectively, these advances demonstrate that progress in biosensing depends not only on the discovery of new sensing mechanisms, but also on continued innovation in materials science and surface engineering to overcome the intrinsic limitations and trade-offs of each sensing platform.
Enzyme-based electrochemical biosensors
Enzyme-based electrochemical biosensors represent one of the most widely utilized platforms in bioanalysis, exploiting the high selectivity and catalytic efficiency of enzymes to convert analytes into electrical signals[135]. These sensors operate through reactions between the enzyme and the analyte. They quantify the oxidation/reduction species generated at the electrode surface[136]. As a result, they offer fast reaction speeds, high sensitivity, and strong selectivity[115,137]. The glucose oxidase-based blood glucose monitoring system is one of the most commercially successful examples of enzyme-based sensors.
Enzyme-based electrochemical biosensors have evolved through several generations[138,139]. Figure 3D illustrates the evolutionary path of enzyme-based electrochemical biosensors. It traces the transition from traditional oxygen-based detection (first generation) to mediator-based electron transfer (second generation) and finally to direct electron transfer between the enzyme and electrode (third generation). The conventional first-generation sensors use oxygen as the electron acceptor and directly measure the generated H2O2. These sensors are straightforward to implement yet remain highly susceptible to interference from changes in oxygen concentration and surrounding contaminants in biological environments. The second-generation sensors introduced redox mediators like ferrocene to stabilize electron transfer, significantly increasing sensitivity. However, the risk of mediator leakage limits their long-term use in biological environments. Third-generation sensors overcame this mediator dependency by enabling direct electron transfer between the enzyme and the electrode. This allows selective reactions at low operating potentials, thereby reducing oxidation/reduction interference and improving both sensitivity and stability. However, challenges such as the precise alignment of the enzyme’s electron transfer site with the electrode complicate sensor design.
Redox-active/non-enzymatic catalytic sensors
Redox-active/non-enzymatic catalytic sensors operate without biological recognition elements like enzymes or aptamers[140]. Instead, they directly oxidize or reduce analytes using catalytic reactions on the electrode surface. Figure 3E illustrates the basic reaction pathway of such non-enzymatic sensors. In this process, the redox pairs of transition metals (e.g., Ni, Co, Cu, Mn) on the electrode surface exchange electrons with the analyte, inducing charge transfer to the electrode[141-143].
This approach presents a straightforward architecture because it does not require biological recognition elements. It also avoids the instability issues associated with protein-based recognition elements such as enzymes, antibodies, and aptamers[144,145]. Notably, metal and metal-oxide nanostructures formed on the electrode surface provide high electrical conductivity and large active surface areas[146]. These features generate significant catalytic currents and support relatively stable long-term operation, making such structures suitable for wearable sensing platforms.
However, the absence of biological recognition elements introduces structural limitations in terms of selectivity[147]. Biofluids contain several metabolites (such as ascorbic acid, uric acid, and glutathione) with similar oxidation potentials, leading to potential interference in electrochemical reactions. Despite these limitations, non-enzymatic sensors offer unparalleled durability, simplicity, low cost, and long-term stability, making them ideal for continuous monitoring of specific metabolites in ISF. Research is ongoing to improve selectivity and reaction speed through electrode compositional control (e.g., tuning transition metal ions, bimetallic alloys, metal-carbon hybrids), crystal facet exposure, and nanostructure engineering[148,149].
Aptamer-based sensors (electrochemical aptamer-based, EAB)
Aptamer-based electrochemical biosensors convert the structural changes that occur when single-stranded DNA (ssDNA) or RNA sequences bind to a target molecule into an electrical signal[150]. Aptamers, which are fixed on the electrode surface, generally have redox reporters such as methylene blue (MB) or ferrocene attached to their ends[151]. Upon binding to the target molecule, the secondary or tertiary structure of the aptamer is rearranged, altering the electron transfer distance, charge distribution, and electron transfer rate between the reporter and the electrode[152]. These structural changes are converted into electrochemical signals such as current, charge transfer resistance, or potential changes. Figure 3F illustrates the typical operating principle of an EAB sensor. In this system, aptamer binding changes the electron-transfer distance between the redox reporter and the electrode, thereby generating a detectable signal.
This reversible sensing mechanism enables real-time and continuous monitoring, unlike antibody-based sensors. Additionally, aptamers can recognize small molecules, hormones, and drugs that lack intrinsic electrochemical activity or are difficult to target using enzyme-based sensing strategies[153,154]. However, aptamer-based sensors often exhibit lower signal amplitudes than enzyme-based sensors. This limitation necessitates signal amplification strategies such as gold nanoparticles, conductive nanosheets, redox cycling, or heterostructure electrodes[155,156]. Furthermore, non-specific protein adsorption, aptamer degradation, and electrode contamination in biological environments can limit long-term stability and signal reproducibility[157].
Antibody-based immunosensors
Antibody-based electrochemical biosensors are diagnostic platforms that convert the classical biological recognition mechanism of antigen-antibody binding into electrical signals. These platforms are widely used for the quantitative analysis of proteins, hormones, and other biomolecules[29]. Antibodies, naturally optimized to bind with high affinity and specificity to target antigens, selectively capture antigens on the electrode surface. This binding alters the charge transfer pathway or the electrochemical characteristics of the surface, leading to measurable signals.
Antibody-based electrochemical immunosensors can be divided into label-free and label-based methods [Figure 3G]. The label-free method (typically EIS-based) directly detects changes in charge transfer resistance and double-layer capacitance induced by antigen binding. This approach eliminates the need for labels or enzymes, offering a simple and stable sensing mechanism. In contrast, the label-based method uses a secondary antibody conjugated with enzymes, metal nanoparticles, or redox mediators, amplifying the current generated after antigen binding[158]. This structure is often implemented in sandwich immunoassays, and catalysts like Cu2O@TiO2-PtCu/Ab2 play a critical role by amplifying the signal through H2O2 reduction[128].
Antibody-based electrochemical sensors achieve unparalleled detection limits and selectivity. In particular, the integration of sandwich structures with nanomaterials establishes the benchmark for high-sensitivity diagnostic technologies[159]. However, due to their rigid protein structure, antibodies do not undergo reversible binding and dissociation. This irreversibility makes continuous monitoring challenging. Additionally, antibodies are sensitive to pH, temperature, and proteolysis, which affects long-term stability[160].
MIP-based electrochemical biosensors
MIP-based electrochemical biosensors use synthetic polymers as recognition elements instead of biological molecules. This bio-free platform enables selective binding by imprinting the structure of a specific analyte into the polymer matrix[16,161]. Figure 3H illustrates the formation process of MIPs on the electrode surface. This process includes the self-assembly of template and functional monomers, electrochemical or chemical polymerization, template removal, and target re-binding.
MIPs are formed by polymerizing the template molecule with functional monomers on the electrode surface, followed by template removal. This process leaves behind binding cavities that mimic the size, shape, charge distribution, and functional group arrangement of the analyte. These cavities provide selective binding like antibodies or aptamers, enabling high specificity. Electrochemical transduction occurs when the target binds to the cavity and changes the electron-transfer pathway or the permeability and ion conductivity of the polymer matrix. These changes are then read out as current, potential, or impedance signals. In EIS, changes in the thickness, density, and permeability of the MIP layer are reflected in the charge transfer resistance (Rct), enabling label-free quantitative detection[162,163].
The key advantage of MIP-based sensors is their structural simplicity, as no biological recognition elements are required. This results in lower manufacturing costs and significantly higher stability under changes in
Electrochemical interference
A critical challenge in applying electrochemical biosensors to complex biofluids, such as whole blood and ISF, is matrix interference originating from endogenous electroactive species. This issue is severely exacerbated when coexisting biomarkers or exogenous drugs have closely overlapping redox potentials[166]. For example, on conventional electrode surfaces, physiological interferents like ascorbic acid (~0.0 V vs. Ag/AgCl), dopamine (~0.2 V), and uric acid (~0.3 V) often exhibit merged oxidation peaks, rendering target resolution nearly impossible[167]. Similarly, detecting H2O2 at relatively high potentials (~0.6 V) in oxidase-based sensors triggers the co-oxidation of lower-potential interferents (~0.4 V), including ascorbic acid, uric acid, and acetaminophen. This co-oxidation inevitably compromises diagnostic accuracy[167].
To systematically mitigate this electrochemical crosstalk, three primary strategies are widely employed across sensing platforms. First, integrating electrocatalytic nanomaterials, such as carbon nanotubes or transition metal oxides, provides differential electrocatalytic activities. This approach artificially widens the peak potential separation, enabling the distinct, simultaneous quantification of overlapping species (e.g., ascorbic acid, dopamine, and uric acid)[168]. Second, the application of permselective membranes serves as a robust physical and electrostatic barrier. Polymeric coatings like Nafion, which are negatively charged, selectively repel anionic interferents such as ascorbic acid and uric acid, preventing them from reaching the electrode interface[169]. Finally, utilizing artificial redox mediators or label-based sensing architectures allows the system to operate at significantly lower or negative potentials. This strategically shifts the detection window away from the highly positive regions where most common endogenous species oxidize, thereby securing the high selectivity required for reliable clinical diagnostics[170].
RECENT ADVANCES IN METABOLITES DETECTION
Glucose
With the rapid growth of the global aging population, diabetes is projected to affect more than 642 million individuals worldwide by 2040[171]. Diabetes is a chronic metabolic disease characterized by insufficient insulin secretion from pancreatic β-cells or impaired hepatic glucose regulation, including disruptions in the glycogen cycle[90,172,173]. Effective diabetes management therefore requires accurate real-time monitoring of glucose levels[173]. Modern CGMs primarily operate based on the enzymatic activity of glucose oxidase (GOx). As illustrated in Figure 4A, GOx catalyzes the oxidation of glucose using the cofactor flavin adenine dinucleotide (FAD), producing gluconic acid while reducing FAD to FADH2. Depending on the generation of the sensor, FADH2 transfers electrons to oxygen (first generation), an electron mediator (second generation), or directly to the electrode surface (third generation). These electron-transfer pathways generate distinct electrochemical outputs, including H2O2 in first-generation sensors and reduced mediator species (Med-red) in second-generation sensors, while third-generation sensors rely on direct electron transfer to the electrode[174].
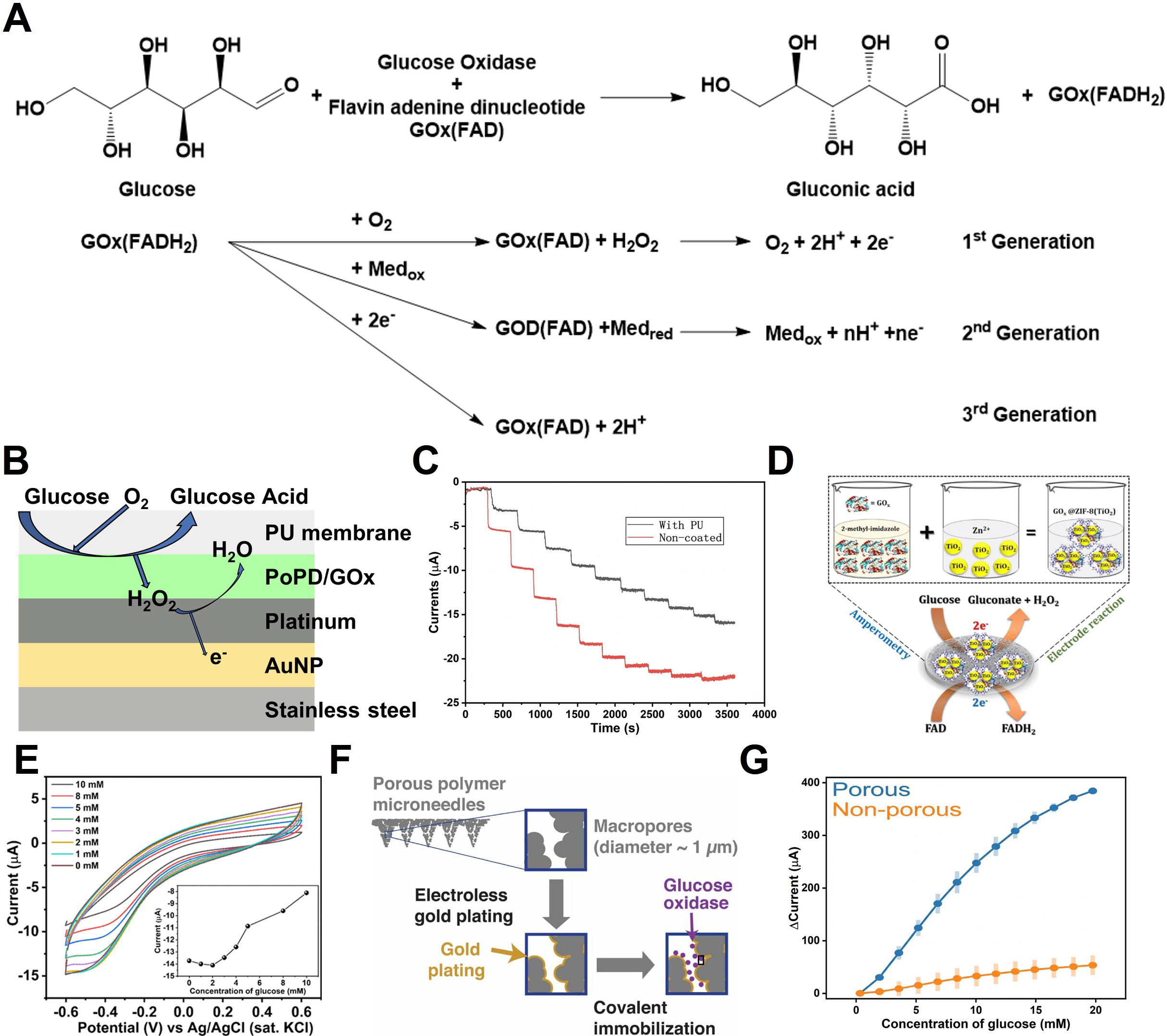
Figure 4. Glucose sensing mechanisms and MN-based electrochemical detection. (A) Enzymatic reaction scheme for glucose sensing. The catalytic oxidation of glucose involves redox cycling that generates a measurable electrochemical current; (B) Schematic illustration of multilayer MN-based glucose sensing; (C) Continuous monitoring performance obtained by CA, comparing MNs with and without a PU encapsulation layer; (D) Synthesis route and sensing mechanism of the GOx@ZIF-8(TiO2) glucose sensor; (E) CV measured at pH 7 for varying glucose concentrations; the inset shows the calibration plot based on cathodic peak currents; (F) Schematic of glucose sensing using porous polymer MNs; (G) Chronoamperometric current responses demonstrating sensitivity variations as a function of MN surface porosity. The error bar in (G) represents the standard deviation. (B and C) Reproduced with permission Copyright 2024, Microchemical Journal[114]. (D and E) Reproduced with permission Copyright 2018, ACS Omega[126]. (F and G) Reproduced with permission Copyright 2021, Journal of Physics: Energy[132]. MN: Microneedle; MNs: microneedles; PU: polyurethane; GOx: glucose oxidase; ZIF: zeolitic imidazolate framework; CV: cyclic voltammetry.
Figure 4B shows the structure of an implantable glucose sensor fabricated through surface functionalization of a stainless-steel acupuncture needle (AN)[114]. The POPD/GOx layer, integrated with Au/Pt nanoparticle (Au/PtNP) coating, simultaneously enhances enzymatic turnover and electrochemical oxidation kinetics. Glucose is oxidized on the GOx-modified electrode surface, producing gluconolactone and H2O2. The generated H2O2 is subsequently oxidized at the PtNP layer, donating electrons to WE. As shown in the chronoamperometric results in Figure 4C, increasing glucose concentrations yield increased formation of H2O2 and, consequently, enhanced oxidation currents. The incorporation of a polyurethane (PU) diffusion membrane decreased the response rate but preserved excellent linearity (R2 = 0.985) across 0-20 mM glucose. Excessively rapid sensor responses can deplete local oxygen and thereby impair linearity in physiological environments. In this context, a diffusion barrier can serve as an effective strategy to mitigate these limitations and achieve more reliable sensor performance.
Metal-organic frameworks (MOFs) offer unique advantages for electrochemical sensors due to their high surface area, tunable porosity, and suitability as enzyme immobilization matrices[175]. Paul et al. developed a GOx@ZIF-8(TiO2) composite by incorporating GOx and TiO2 nanoparticles into a ZIF-8 framework to enhance glucose oxidation kinetics [Figure 4D][126]. The imidazolate groups of ZIF-8 promote uniform enzyme immobilization via hydrophobic interactions and hydrogen bonding. The composite was drop-cast onto a glassy carbon electrode (GCE), and current responses were recorded [Figure 4E]. The H2O2 generated by enzymatic oxidation suppressed oxygen reduction at the cathode, resulting in a decrease in cathodic current. The sensor exhibited tandem catalytic activity toward both glucose oxidation and H2O2 reduction, enabling glucose detection over a 1-10 mM range with a detection limit of 80 nM.
Similarly, reduced graphene oxide (rGO) has been widely employed as a conductive two-dimensional platform to enhance non-enzymatic electrochemical glucose sensing. In hybrid systems such as ZnO-CeO2/rGO, rGO effectively compensates for the limited electrical conductivity of metal oxides by facilitating rapid charge transfer and reducing interfacial resistance[176]. In addition, its planar nanosheet structure promotes uniform dispersion of catalytic nanoparticles and increases accessible active sites, leading to enhanced electrocatalytic activity. As a result, the composite sensor achieved a high sensitivity of
Another study demonstrated a high-performance glucose sensor using a gold-coated porous polymer MNA in a three-terminal configuration [Figure 4F][132]. The system consisted of an Au/GOx/Poly(VF-co-HEMA) WE, Au CE, and Ag/AgCl RE. GOx was immobilized using cystamine-modified Au surfaces, while Poly(VF-co-HEMA) served as an electron mediator to reduce oxygen and H2O2-dependent variations. CA at +0.3 V (vs. Ag/AgCl) in PBS revealed that the nonporous MN exhibited a sensitivity of 3.16 μA/mM, whereas the porous MN achieved 22.99 μA/mM [Figure 4G][132]. However, the porous structure introduced a longer response time (~82 s) due to diffusion delays within the pore network. The mediator-based second-generation mechanism employs Med/Med+ to facilitate electron transfer directly between GOx and the electrode. This bypasses H2O2 formation and thereby minimizes interference. This method is more stable because it does not depend on dissolved oxygen concentration and enables low-potential CGM operation.
Lactate
Blood lactate concentration acts as a critical biomarker for tissue oxygenation and metabolic status. It provides essential prognostic value in clinical emergencies, including sepsis, shock, and systemic hypoperfusion[30,31,177,178]. Under low-oxygen conditions, pyruvate is reduced to lactate by lactate dehydrogenase (LDH). This reaction oxidizes NADH to regenerate NAD+, sustaining ATP production under hypoxic stress [Figure 5A-1][179,180]. The generated lactate is subsequently reconverted to pyruvate for gluconeogenesis in the liver or directly oxidized as a fuel source in cardiac and skeletal muscle[181]. Consequently, elevated lactate levels reflect systemic hypoperfusion or mitochondrial dysfunction.
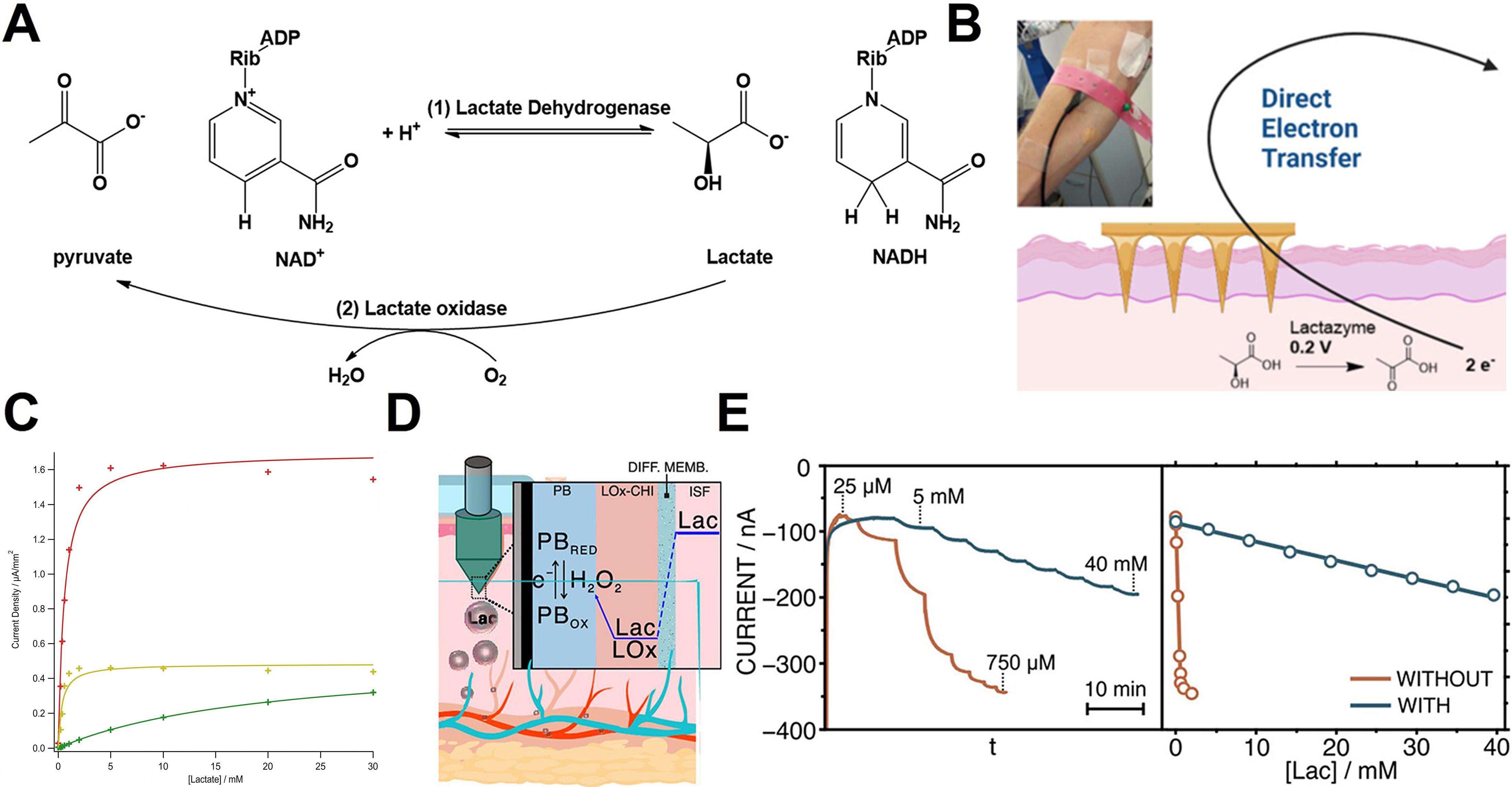
Figure 5. Enzyme-based lactate sensing mechanisms and applications. (A) Enzymatic reaction scheme for lactate sensing; (B) Lactazyme-immobilized MN lactate sensor; (C) Current response as a function of acetate-cellulose coating thickness; (D) Mechanism of ISF-based lactate sensing. H2O2 generated by lactate oxidase undergoes spontaneous redox cycle with PB, producing a detectable current; (E) Effect of a hydrated polymer layer on MN surfaces. (B and C) Reproduced with permission Copyright 2023, ACS Sensors[182]. (D and E) Reproduced with permission Copyright 2024, ACS Sensors[127]. MN: Microneedle; ISF: interstitial fluid; PB: Prussian blue; ADP: adenosine diphosphate; NAD+: nicotinamide adenine dinucleotide (oxidized form); NADH: nicotinamide adenine dinucleotide (reduced form); LOx: lactate oxidase; CHI: chitosan; PBRED: reduced Prussian blue; PBOX: oxidized Prussian blue; Lac: lactate; DIFF. MEMB: diffusion membrane.
Electrochemical sensing strategies emulate these biochemical pathways. LDH-based sensing detects the electrochemical oxidation of NADH generated when lactate is enzymatically converted to pyruvate. Alternatively, lactate oxidase (LOx)-based approaches exploit the LOx-mediated oxidation of lactate to pyruvate and H2O2 [Figure 5A-2]. Despite its dependence on dissolved oxygen, LOx-based detection remains widely used in clinical and wearable sensing due to its high selectivity and straightforward electrode design. Through these mechanisms, lactate’s redox transformations are efficiently transduced into electrical signals, enabling real-time monitoring of metabolic status.
Figure 5B presents a MN-based lactate sensor designed to overcome the potential and selectivity limitations of conventional H2O2-dependent systems by leveraging a third generation enzyme sensor architecture[182]. Polycarbonate MNA was fabricated via injection molding, followed by metallization with a Ti/Au or Ti/Ag coatings to form the WE, CE, and Ag/AgCl RE. Lactazyme was immobilized by drop-casting a 25 μL mixture of enzyme and carbon ink onto the gold-coated MNs, producing a conductive bioelectrocatalytic film. A cellulose acetate diffusion-limiting membrane was applied to regulate analyte flux, protect the enzyme layer, and extend the linear operating range.
The sensor operates via direct electron tunneling from the heme-domain active site of lactazyme to the electrode at 0.2 V, minimizing interference from endogenous redox molecules. As shown in Figure 5C, high current densities and rapid saturation occur at 5 mM lactate without the diffusion barrier. In contrast, a 3% cellulose acetate layer reduces the current amplitude by approximately fivefold while extending the linear detection range beyond 10 mM[182]. This helps prevent enzyme overload and supports accurate monitoring across physiological to supraphysiological lactate concentrations (1-20 mM).
Figure 5D illustrates a second-generation electrochemical sensor that integrates lactate oxidase with an optimized diffusion-limiting membrane for wide-range, real-time measurement in skin ISF[127]. The WE was constructed by coating a carbon layer onto a stainless-steel MN, followed by 20 CV cycles between -0.5 and 0.6 V. A chitosan-lactate oxidase (CHI-LOx, 0.5 μL) mixture was then immobilized onto the electrode. A doped PVC membrane was subsequently coated to regulate lactate flux and stabilize the Prussian blue (PB) redox state during operation at -0.1 V.
The sensing mechanism proceeds through LOx-mediated oxidation of lactate to H2O2, followed by spontaneous PB reduction. This process generates a faradaic current proportional to lactate concentration. As shown in Figure 5E, the membrane dramatically improves the linear range: without the membrane, the sensor saturates at lactate concentrations of 0.1-0.75 mM, whereas with the membrane, the dynamic range extends to 0.25-40 mM. This broad range is necessary given the wide variation in lactate levels across physiological and pathological conditions [normal ISF (0.5-1.5 mM), hyperlactatemia (3-5 mM), and strenuous anaerobic exercise (> 15 mM)].
Creatinine
Creatinine is generated endogenously through the non-enzymatic cyclization of creatine and phosphocreatine and serves as a critical metabolic indicator reflecting both muscle mass and renal function[183,184]. Because creatinine is cleared almost exclusively by glomerular filtration, its concentration in blood is a central biomarker for diagnosing acute kidney injury, chronic renal failure, glomerulonephritis, and dehydration[185-187]. Enzyme-based creatinine sensing typically follows one of two biochemical pathways[188]. In the first, creatinine is converted to N-methylhydantoin and NH3 by creatinine deiminase [Figure 6A-i]. In the second, creatinine is hydrolyzed to creatine by creatinine amidohydrolase [Figure 6A-ii], followed by conversion to urea and sarcosine via creatine amidinohydrolase [Figure 6A-iii]. The resulting urea and sarcosine are subsequently degraded by urease, sarcosine oxidase, and glycine oxidase to produce NH3 and H2O2[189].
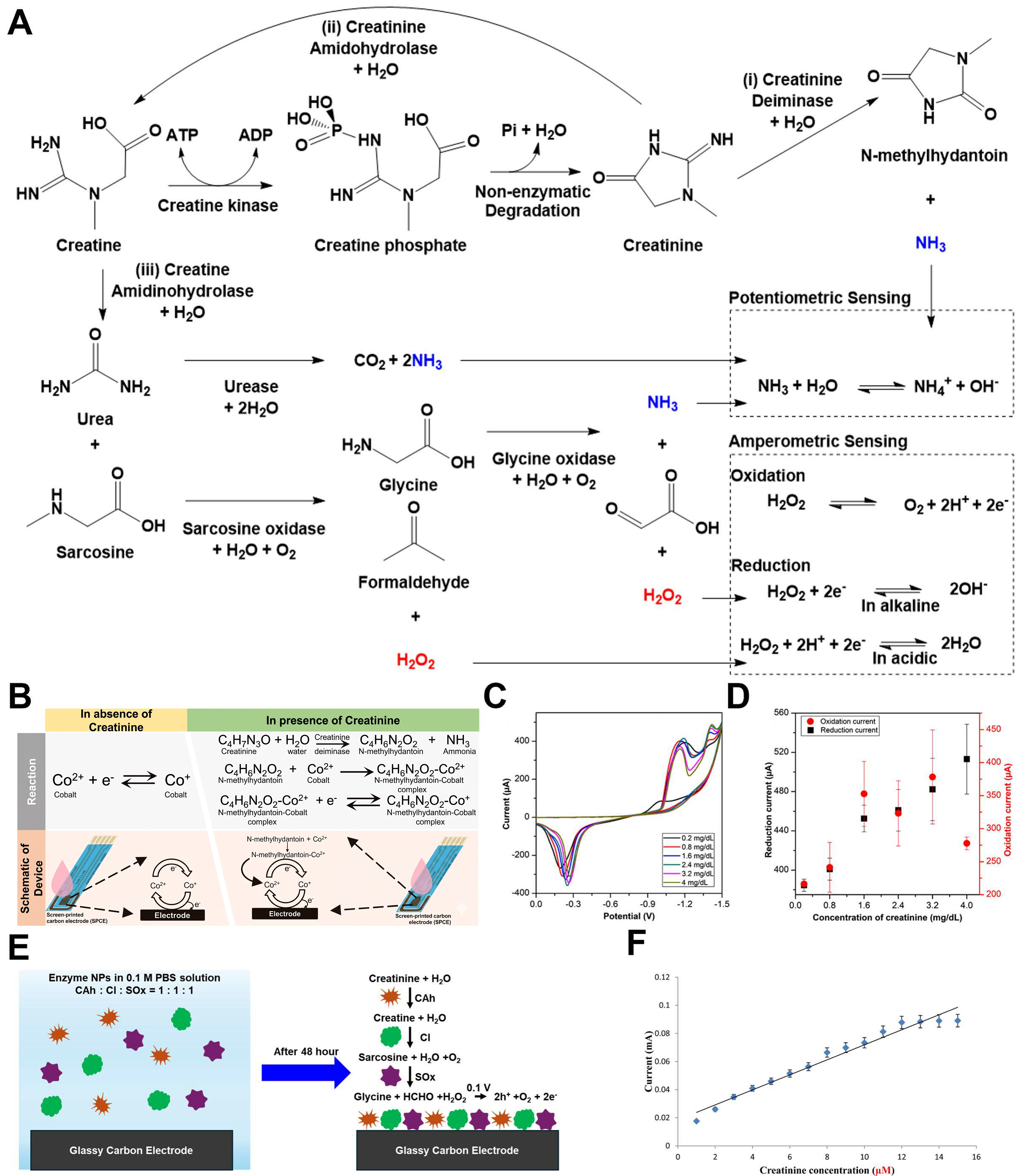
Figure 6. Creatine metabolic pathway and creatinine sensing mechanisms. (A) Creatine undergoes non-enzymatic degradation to spontaneously form creatinine, which is subsequently decomposed through enzymatic reactions to yield electroactive species. The generated H2O2 can undergo either oxidation or reduction; (B) Mechanism of a second-generation enzyme-based creatinine sensor. H2O2 produced from the enzymatic cascade modulates the oxidation state of Co; (C) CV obtained at varying creatinine concentrations; (D) Calibration plots of current versus concentration extracted at -1.4 V (reduction) and +1.4 V (oxidation); (E) Fabrication scheme and enzymatic reaction pathway of the CAhNP/CINPs/SOxNPs/GCE sensor; (F) Current-concentration plots obtained from CV (-0.3 to
Electrochemical monitoring translates the NH3 and H2O2 generated during these reactions into measurable electrical outputs via potentiometry or amperometry[92]. Potentiometric sensors quantify creatinine by monitoring NH3-induced potential shifts governed by the NH4+/OH- equilibrium. However, their sensitivity is affected by pH and temperature variations. Amperometric sensors provide a current linearly proportional to analyte concentration with rapid response times, making them more appropriate for real-time, wearable monitoring[190].
As noted above, creatinine sensing often necessitates multi-enzyme cascades to generate detectable quantities of H2O2 or NH3. This reliance on complex enzymatic pathways not only increases fabrication costs but also heightens measurement uncertainty. Moreover, direct electrochemical oxidation of NH3 produced by single-enzyme pathways (e.g., creatinine deiminase alone) requires high overpotentials (> 0.8 V vs. Ag/AgCl), making the signal highly susceptible to electrode fouling and unsuitable for continuous amperometric operation.
Figure 6B illustrates the second-generation sensor in which creatinine deiminase converts creatinine to N-methylhydantoin[191]. The resulting N-methylhydantoin subsequently forms complexes with transition metal ions to generate a redox signal. Cobalt ions, in particular, interact strongly with N-methylhydantoin and undergo immediate redox reactions at the electrode surface, amplifying the current response. As shown in Figures 6C and D, the reduction current measured at -1.4 V showed a linear correlation with creatinine concentration. Nevertheless, the requirement for cobalt chloride solution and the high reduction potential
Figure 6E presents the fabrication of an amperometric sensor employing a multi-enzyme creatinine-creatine-sarcosine cascade[125]. Creatinine amidohydrolase (CAh), creatine amidinohydrolase (CI), and sarcosine oxidase (SOx) were immobilized as enzyme nanoparticles (ENPs) by crosslinking with cysteamine dihydrochloride. An activated glassy carbon electrode was immersed in the ENP dispersion to construct an ENP/GC sensor. Upon reaching the electrode surface, creatinine undergoes sequential CAh-CI-SOx conversion, ultimately producing H2O2, which is detected amperometrically. The calibration curve for creatinine concentration was generated from CV measurements acquired between -0.3 and 1 V [Figure 6F]. The sensor achieved a wide detection range (0.01 μM-12 mM), effectively covering the full physiological spectrum of creatinine - from normal levels (45-140 μM) to concentrations exceeding 1 mM in renal failure. This confirms that the tri-enzyme cascade effectively avoids saturation, even at high concentrations.
Urea
Blood urea nitrogen (BUN) serves as a key clinical indicator for the accumulation of nitrogenous end-products. BUN levels are influenced by various pathological states, such as acute kidney injury, heart failure, and liver dysfunction[192-194]. Urea is synthesized in the liver through the urea cycle, which serves as the principal detoxification pathway converting NH3 into urea [Figure 7A][195]. In hepatic mitochondria, carbamoyl phosphate synthetase I (CPS I) converts NH3 and CO2 into carbamoyl phosphate[196]. Ornithine transcarbamylase (OTC) then combines carbamoyl phosphate with ornithine to form citrulline. Argininosuccinate synthase (ASS) then incorporates aspartate to generate argininosuccinate, which is cleaved to form arginine and fumarate. Arginase catalyzes the hydrolysis of arginine to yield urea and ornithine, with the latter re-entering the mitochondria to sustain the cycle[197]. The urea produced circulates systemically and is filtered and excreted by the renal glomeruli. Therefore, blood urea concentrations reflect both hepatic synthesis and renal elimination.
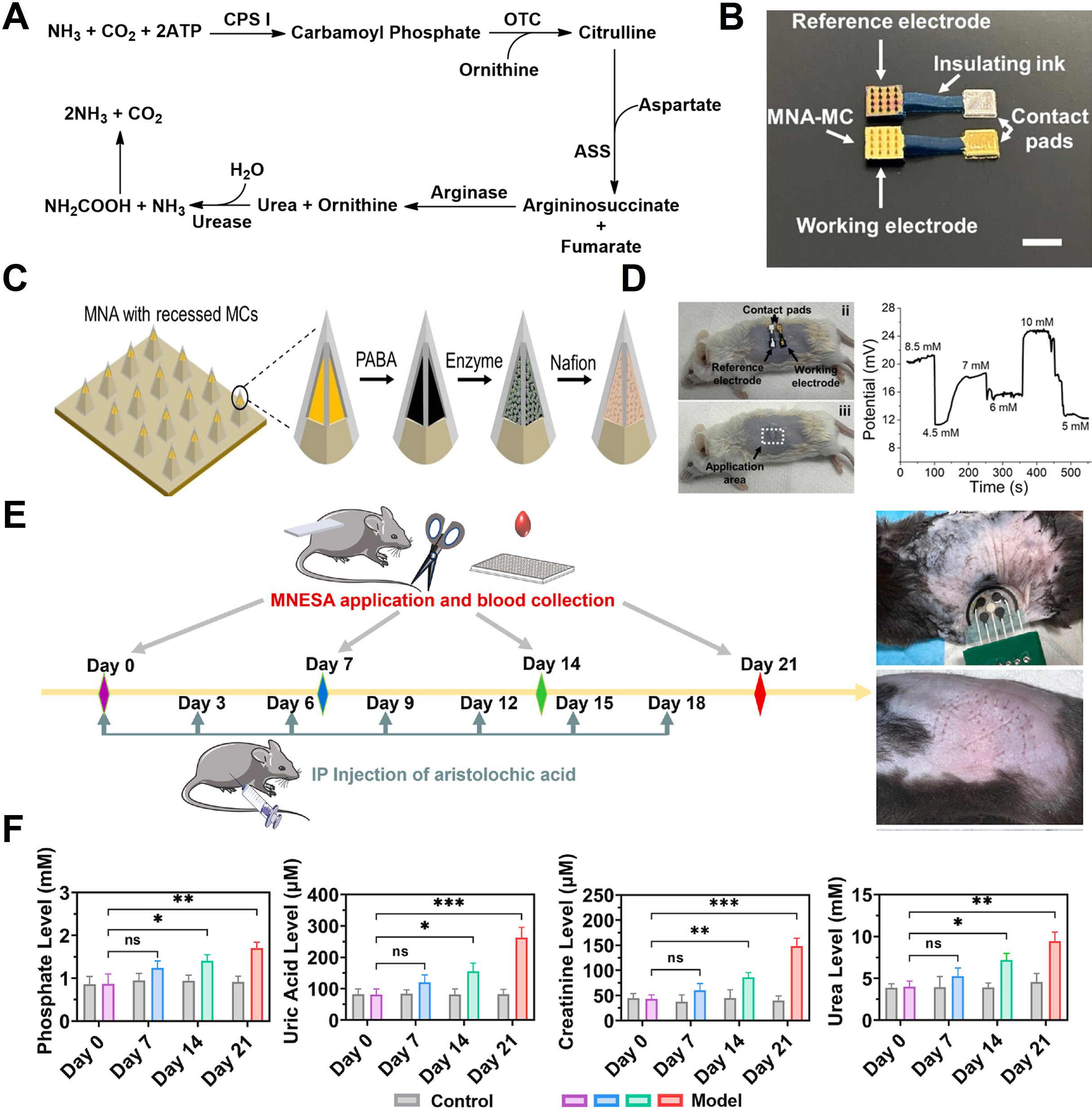
Figure 7. Urea metabolic pathway and sensing mechanisms. (A) Flow map of enzyme-based detection strategies for urea generated through the urea cycle; (B) Schematic of a solid MN designed for urea sensing. The MN incorporates an embedded microcavity architecture composed of a PABA/enzyme/Nafion stack; (C) Optical images of ex-vivo tests; (D) Experimental protocol for in vivo evaluation of an integrated sensor capable of measuring phosphate, uric acid, creatinine, and urea; (E) Temporal concentration profiles of each metabolite before (grey) and after administration of aristolochic acid (pink, blue, green, red); (F) Phosphate, uric acid, creatinine, and urea levels in ISF of control and comparison mouse. Each data point represents the mean and standard error of three measurements taken for a single mouse model. All concentration values were calibrated against in-vitro measurements. The error bar in (F) shows the standard deviation. (B-D) Reproduced with permission Copyright 2024, ACS Sensors[199]. (E and F) Reproduced with permission Copyright 2023, Biosensors and Bioelectronics[124]. *P < 0.05, **P < 0.01, ***P < 0.001. MN: Microneedle; PABA: polyaniline-boronic acid; ATP: adenosine triphosphate; CPS I: carbamoyl phosphate synthetase I; OTC: ornithine transcarbamylase; ASS: argininosuccinate synthase; MNA: microneedle array; MC: microcavity; MCs: microcavities; MNESA: MN electrical sensor array; IP: intraperitoneal; ns: not significant.
Electrochemical BUN sensing commonly employs urease, which hydrolyzes urea to NH3 and CO2, resulting in local pH elevation and an increase in NH4+ concentration. These changes in the NH3/NH4+ equilibrium drive measurable potential shifts in potentiometric sensors, enabling quantification through ammonium-selective electrodes[198]. Figure 7B presents an optical micrograph of an enzyme-immobilized MN sensor incorporating recessed microcavities[199]. A polyaniline-boronic acid (PABA) film was first electropolymerized onto a gold-coated MN, serving both as an enzyme-immobilization matrix and a potentiometric transducer [Figure 7C]. Following urease immobilization, the electrode was encapsulated by Nafion. The Nafion layer markedly enhanced sensor durability, retaining 92% of its initial performance after 10 days of storage in PBS (compared with a 95% loss after 4 days without encapsulation). The sensing mechanism arises from local deprotonation interactions between NH3/NH4+ and the PABA film, which manifest as shifts in the open-circuit potential. The device was evaluated ex vivo using euthanized mouse skin [Figure 7D]. Artificial ISF containing 4-10 mM urea was injected subcutaneously, and the wearable patch achieved a relative standard deviation of 4.7%-8.4%. These results demonstrate that the multilayer geometry effectively protected the sensing layer during skin insertion while enabling stable detection.
Given that many pathological conditions simultaneously alter various metabolites, simultaneous monitoring of multiple biomarkers is essential to avoid diagnostic inaccuracy. Renal dysfunction not only affects traditional renal markers but also alters metabolites such as phosphate, uric acid, creatinine, and urea. Although numerous sensors have been developed for individual analytes, single-biomarker detection can be misleading. For example, dehydration elevates both creatinine and urea, diabetes increases circulating uric acid, and hypertension increases phosphate levels. To address this challenge, in vivo evaluation of an enzyme-functionalized polymer MN-based multi-biomarker sensing system was conducted [Figure 7E][124]. A chronic renal failure model was induced in mice via intraperitoneal administration of aristolochic acid every three days. For continuous monitoring over 21 days, an MN electrical sensor array (MNESA) was applied to the dorsum. This array consisted of four WEs functionalized with uricase, CAh/CI/SOx, urease, and ammonium heptamolybdate, alongside an Ag/AgCl RE. As shown in Figure 7F, the concentrations of all four biomarkers increased over time. One-way ANOVA revealed no statistically significant changes on day 7, whereas two-way ANOVA incorporating all biomarkers detected significant differences (P < 0.05). These results demonstrate that multi-analyte sensing can improve the early diagnostic sensitivity of kidney disease by capturing the interconnected metabolic changes in the body.
RECENT ADVANCES IN HORMONE DETECTION
Cortisol
Cortisol is a key regulatory factor in the HPA axis, playing a central role in various physiological processes, including metabolic balance, immune function modulation, and stress adaptation[98,200]. Abnormal cortisol levels are closely associated with several health conditions, such as Cushing’s syndrome, Addison’s disease, post-traumatic stress disorder (PTSD), major depressive disorder, chronic fatigue syndrome, as well as metabolic and autoimmune diseases[201,202]. Free cortisol, which is not bound to proteins, is biologically active and distributed in biofluids such as sweat, saliva, and ISF, passing through capillaries. Consequently, non-invasive biofluid-based cortisol monitoring has emerged as a critical tool for stress physiology, mental health evaluation, and endocrine disease diagnosis. In particular, the ability to track diurnal fluctuations of cortisol concentrations (20-400 nM) in real-time using precise electrochemical sensor technologies is gaining significant importance[203].
Yeasmin et al. introduced an electrocatalytic molecularly imprinted polymer (EC-MIP) strategy for highly selective cortisol detection[133]. Their approach involves doping polypyrrole (PPy) with Cu phthalocyanine tetrasulfonate (CuPcTS), followed by electrochemical polymerization with cortisol as a template. This process creates a molecular recognition cavity on the electrode surface [Figure 8A]. After template removal, the imprinted cavity selectively binds to reintroduced cortisol because it retains complementary structural and functional group arrangements. This binding activates the electrocatalytic reaction by modulating electron transfer through CuPcTS. As shown in Figure 8B, cortisol binding promotes the Cu2+/Cu+ redox transition, enhancing the electrocatalytic hydrogenation pathway, thereby leading to increased current signals. This catalytic amplification mechanism provides a signal-on response that is sensitive to concentration changes, with amperometric results in Figure 8C clearly showing a marked increase in current with rising cortisol concentrations. In contrast, the unprinted PPy electrode exhibited minimal concentration dependence, highlighting the significant role of the imprinting structure.
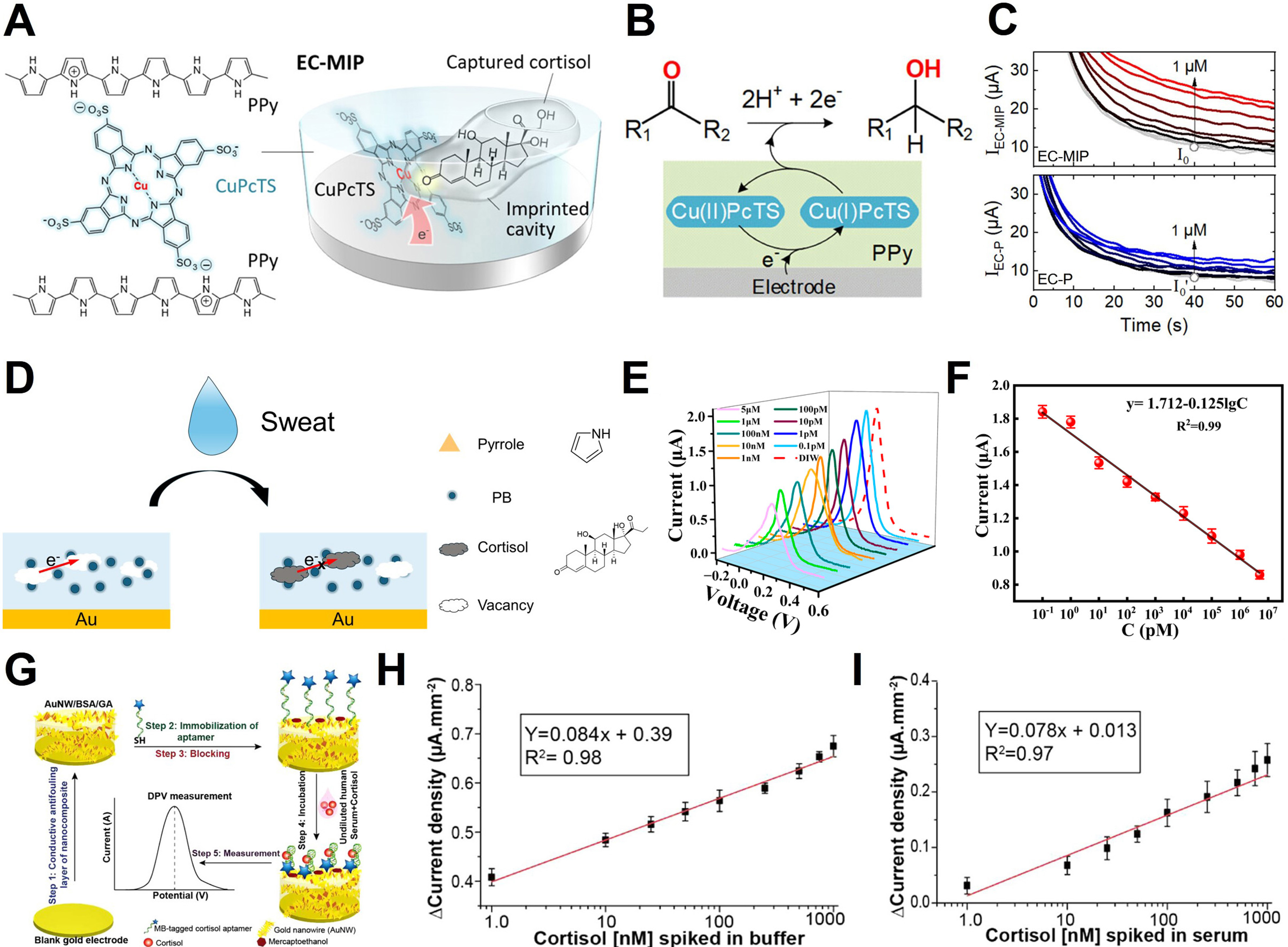
Figure 8. Cortisol sensing mechanism and applications. (A) Structure of the EC-MIP sensor for cortisol detection, using CuPcTS-doped polypyrrole (PPy); (B) Electrocatalytic hydrogenation mechanism at the EC-P electrode; (C) Amperometric responses of the sensor at cortisol concentrations (0 to 1 μM). (Top: EC-MIP sensor, showing a clear increase in current; Bottom: EC-P electrode, showing a less responsive current); (D) Schematic representation of the cortisol detection process using the MIP@PI sensor; (E) DPV current responses of the MIP@PI sensor at various cortisol concentrations; (F) Calibration curve of the MIP@PI sensor; (G) Schematic overview of the aptasensor fabrication process: (Step 1) Conductive antifouling layer synthesis, (Step 2) Aptamer immobilization, (Step 3) Blocking, (Step 4) Incubation with sample, (Step 5) DPV measurement for cortisol detection; (H) DPV current density response to varying cortisol concentrations spiked in buffer; (I) DPV current density response to varying cortisol concentrations spiked in undiluted human serum. The error bars in (F), (H) and (I) represent the standard deviation. (A-C) Reproduced with permission Copyright 2023, ACS Applied Materials & Interfaces[133]. (D-F) Reproduced with permission Copyright 2025, Biosensors[35]. (G-I) Reproduced with permission Copyright 2021, ACS Omega[36]. EC: Electrocatalytic; MIP: molecularly imprinted polymer; PI: polyimide; DPV: differential pulse voltammetry; PB: Prussian blue; MB: methylene blue.
Chen et al. developed a Polyimide (PI)-based molecularly imprinted polymer (MIP@PI) platform for cortisol detection, focusing on stability in wearable environments[35]. This approach uses synthetic polymers, without biomolecular recognition elements, to polymerize pyrrole-Prussian blue (Py-PB) and cortisol simultaneously on the electrode surface. After template removal, cortisol-specific cavities are formed [Figure 8D]. When cortisol rebinds to the cavity, electron transfer via PB nanoparticles is inhibited, resulting in a signal-off response observed in DPV measurements, with the current decreasing in a concentration-dependent manner [Figure 8E]. The calibration curve in Figure 8F shows strong linearity (R2 = 0.99) between current decrease and cortisol concentration, demonstrating the platform’s high quantitative reliability. This platform exhibits excellent resistance to environmental interference, including heat, enzyme degradation, and protein fouling, making it especially advantageous for sweat-based cortisol monitoring.
Singh et al. constructed an electrochemical aptasensor designed to undergo a conformational change upon cortisol binding[36]. The core of this platform is a conductive antifouling layer based on Au nanowires, bovine serum albumin (BSA), and glutaraldehyde (GA) assembled on a gold electrode. This layer prevents nonspecific protein adsorption and stabilizes the signal generated by aptamer conformational changes [Figure 8G]. Upon cortisol binding, the electron transfer efficiency of the redox reporter, MB, changes, which is read directly through DPV [Figure 8H]. The DPV current density increases linearly with rising cortisol concentrations, demonstrating excellent quantitative performance (R2 = 0.98). Furthermore, Figure 8I demonstrates that the sensor maintains this linearity even in undiluted human serum (R2 = 0.97), indicating the high reproducibility and selectivity of the antifouling nanocomposite-based electrode in high-protein biological fluids.
Insulin
Insulin is a polypeptide hormone secreted by the beta cells of the pancreas, playing a critical role in regulating glucose absorption and metabolism in the body[204,205]. More broadly, insulin regulates the metabolism of carbohydrates, fats, and proteins and helps maintain normal blood glucose levels[206]. It promotes the absorption of glucose into liver, muscle, and adipose tissues, where it is converted into energy. Dysregulation of insulin secretion is closely linked to both Type 1 and Type 2 diabetes[207]. In these conditions, insulin secretion or insulin responsiveness is impaired, leading to abnormal blood glucose levels and potential long-term complications.
Dervisevic et al. reported the first insulin biosensor based on a high-density polymer MNA modified with insulin-specific aptamers, designed for transdermal monitoring of insulin levels in ISF[25]. In this study, the sensor extracts insulin from ISF using the MNA. The extracted insulin then binds to insulin-specific aptamers immobilized on the electrode surface, and the resulting electrochemical signal is measured. Figure 9A illustrates the operational principle of this aptasensor, where the binding between insulin and aptamers is detected via EIS. Both in vitro and in vivo experiments demonstrated that the MNA-based insulin biosensor successfully extracts and electrochemically monitors insulin levels in ISF.
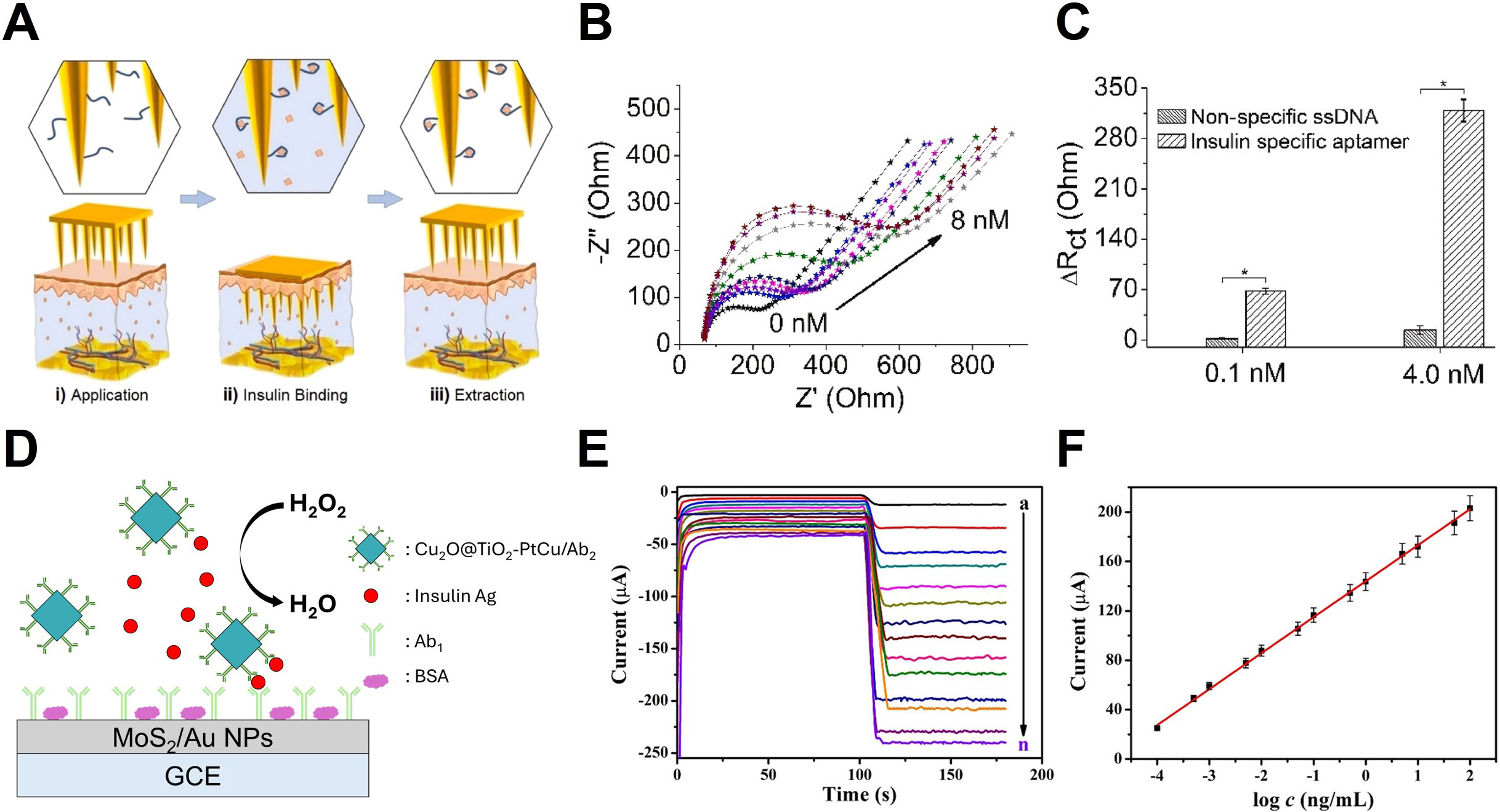
Figure 9. Microneedle base electrochemical insulin sensing. (A) Steps involved in aptasensor operation: (i) Extraction of insulin from ISF, (ii) binding of the extracted insulin to the immobilized aptamer, and (iii) EIS; (B) Electrochemical response of the aptasensor during cortisol detection; (C) Comparison of ∆Rct (change in charge-transfer resistance) for insulin-specific aptamers and non-specific ssDNA.
Figure 9B presents the electrochemical response of the MNA-based aptasensor. The experiment measured the EIS of the electrode after incubation with a range of insulin concentrations (0.01 to 8 nM) in artificial ISF (aISF). The results showed that the impedance (Z″) changed as the insulin concentration increased, indicating that insulin binding to the aptamer altered the charge-transfer properties of the electrode. This change was proportional to concentration, with higher insulin concentrations leading to greater shifts in impedance.
Figure 9C shows a histogram comparing the change in charge transfer resistance (ΔRct) values obtained from EIS measurements. The comparison between ssDNA-modified electrodes (control) and insulin-specific aptamer-modified electrodes revealed that the ssDNA electrodes showed minimal response at insulin concentrations of 0.1 and 4.0 nM. Conversely, the insulin-specific aptamer modified electrodes exhibited clear impedance changes in response to varying insulin concentrations. This demonstrates the high selectivity of the MNA-based aptasensor for insulin, minimizing errors from non-specific binding.
Li et al. developed a sandwich structure immunosensor for insulin antigen (Ag) detection, which demonstrates high performance[128]. Figure 9D explains the concept of the sandwich structure immunosensor using MoS2/Au NP-modified GCE. In this structure, insulin is captured through antigen-antibody binding, and the Cu2O@TiO2-PtCu/Ab2 nanocomposite acts as a catalytic label to amplify the electrochemical signal. This catalytic system enhances the electrochemical signal by accelerating H2O2 decomposition, resulting in larger current changes with increasing insulin concentrations and thereby improving sensor sensitivity and reliability.
Figure 9E presents the amperometric responses of the immunosensor over a broad concentration range of insulin Ag. The current signal rose proportionally with insulin concentrations from 0.1 pg/mL to 100 ng/mL, reflecting the increased density of Cu2O@TiO2-PtCu/Ab2 labels on the electrode surface. Because these labels catalyze H2O2 reduction, higher antigen binding yields more active catalytic sites, thereby amplifying the measured current. The resulting concentration-dependent current profiles confirm that the immunosensor provides a robust and quantitative electrochemical response over a broad dynamic range. Figure 9F presents the calibration curve of the sensor, showing a linear relationship between amperometric response and insulin Ag concentration. The regression equation for the calibration curve was I = 29.14 logC + 144.08, with a correlation coefficient (R2) of 0.9985, indicating high precision. The detection limit was calculated as
RECENT ADVANCES IN PHARMACOLOGICAL MOLECULES
Vancomycin
Vancomycin is a glycopeptide antibiotic widely used as a “last-resort” treatment for serious Gram-positive bacterial infections, including methicillin-resistant Staphylococcus aureus (MRSA)[208,209]. Despite its clinical significance, vancomycin is characterized by a narrow therapeutic window[210]. Sub-therapeutic levels can lead to treatment failure and the emergence of drug-resistant strains, such as vancomycin-resistant Enterococci (VRE)[211]. Conversely, excessive dosages are associated with severe adverse effects, including nephrotoxicity and ototoxicity[211,212]. Furthermore, the pharmacokinetics of vancomycin vary significantly among individuals depending on renal function, necessitating precise dosage adjustments Therefore, therapeutic drug monitoring (TDM) is essential to maximize therapeutic efficacy while minimizing toxicity. This creates a strong need for sensing platforms capable of rapidly and accurately quantifying vancomycin concentrations in clinical settings.
Figure 10A provides a comprehensive overview of the pharmacokinetics and molecular mechanism underlying vancomycin-induced inhibition of Gram-positive bacterial cell wall synthesis. As illustrated in the schematic pathway, intravenously administered vancomycin distributes via the bloodstream to target infection sites. The drug exerts its bactericidal effect by specifically binding to the D-Ala-D-Ala residues of peptidoglycan precursors. This interaction physically prevents the transpeptidase enzyme (shown in green) from catalyzing the essential crosslinking process, ultimately leading to bacterial lysis. The structural basis of this specific inhibition is further elucidated in Figure 10B, which details the high-affinity complex formation between vancomycin and the Ac-L-Lys(Ac)-D-Ala-D-Ala motif. The extensive hydrogen bonding network, highlighted by red dashed lines, induces significant steric hindrance that is critical for blocking the enzymatic access required for cell wall polymerization.
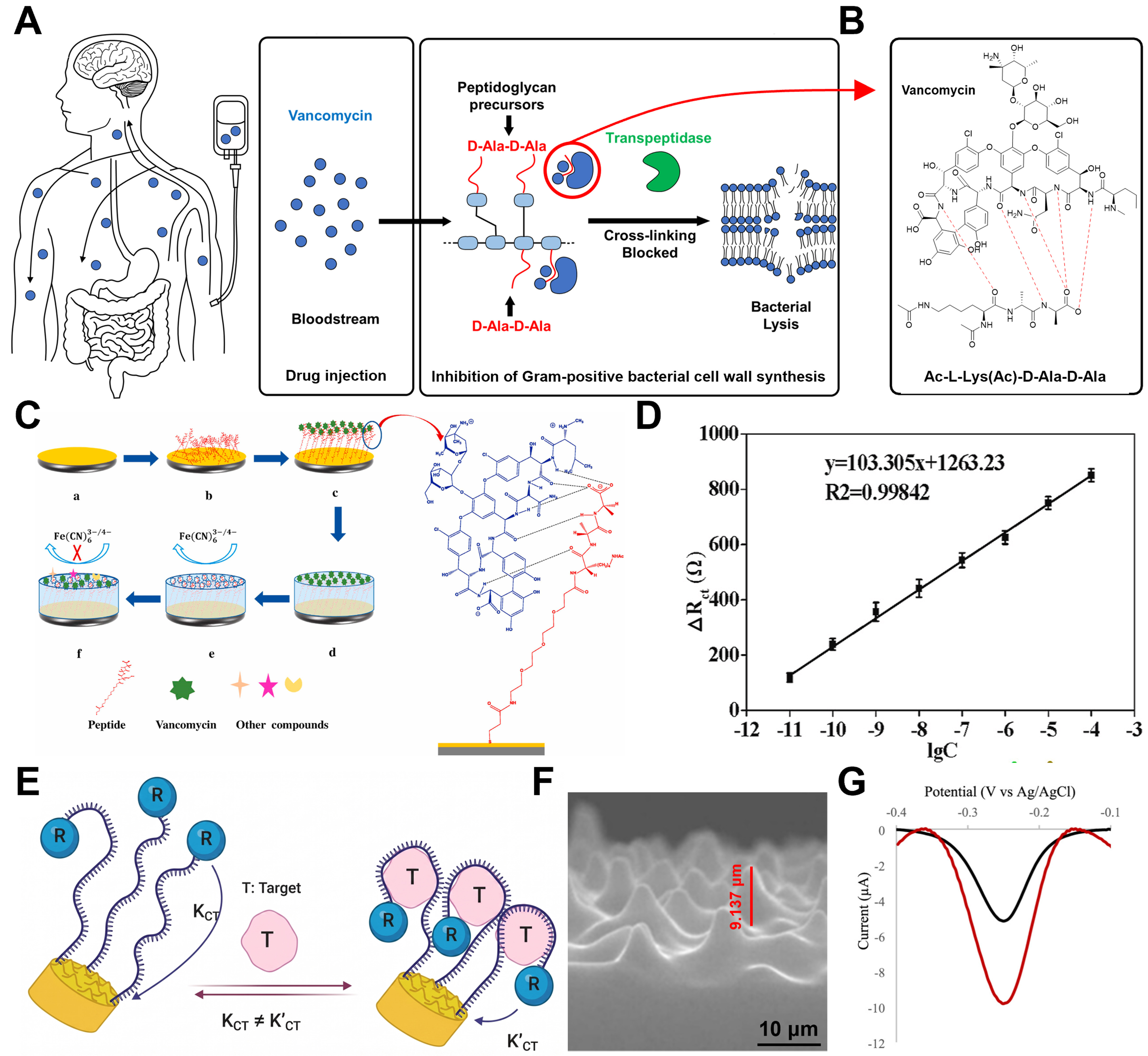
Figure 10. Vancomycin’s therapeutic mechanism for nephritis and sensing devices. (A) Bactericidal mechanism of vancomycin inhibiting cell wall synthesis by specifically binding to the D-Ala-D-Ala terminus; (B) Illustration of the five specific hydrogen bonds formed between vancomycin and the D-Ala-D-Ala terminus11; (C) Sensing principle of the hybrid peptide-MIP sensor, where target capture blocks electron transfer; (D) Linear calibration curve of electron transfer resistance ∆Rct) versus vancomycin concentration (R2 ≈ 0.998); (E) Schematic of the aptamer-based sensor using a 3D MSE for enhanced sensitivity; (F) Cross-sectional SEM image of the 3D MSE; (G) Comparison of SWV responses demonstrating the superior signal gain of the 3D MSE (red) over the planar electrode (black). The error bar in (D) represents the standard deviation. (C and D) Reproduced with permission Copyright 2021, Biosensors and Bioelectronics[134]. (E-G) Reproduced with permission Copyright 2025, ACS Omega[130]. R2: Coefficient of determination; 3D: three-dimensional; MSE: microstructured electrode; SEM: scanning electron microscope; SWV: square wave voltammetry.
Tan et al. proposed a hybrid sensor combining peptide affinity and MIPs for high-sensitivity vancomycin detection[134]. Figure 10C illustrates the fabrication and working principle of this sensor. A polydopamine layer was formed around the vancomycin-peptide complex immobilized on gold nanoparticles, followed by template removal to create specific recognition cavities. The detection is based on the mechanism where the rebinding of vancomycin physically blocks these cavities, hindering the electron transfer of the redox probe [Fe(CN)63-/4-]. As shown in Figure 10D, the change in charge transfer resistance (∆Rct) exhibited a linear relationship with the logarithm of vancomycin concentration. The limit of quantification (LOQ) was as low as 10 pM, providing sufficient sensitivity for real sample analysis even after significant dilution (10-100 times). This represents a distinct analytical advantage, as significant sample dilution helps minimize matrix interference.
Haji-Hashemi et al. presented a 3D microstructured electrode (MSE)-based EAB sensor for non-invasive sweat monitoring[130]. To address the low signal output of planar electrodes, they introduced gold-coated 3D microstructures as shown in Figure 10E. This architecture provides an effective surface area (1.46 cm2) approximately twice that of planar electrodes, enhancing aptamer loading and redox current generation. Figure 10F displays the cross-sectional SEM image of the gold-coated PDMS structure. As evidenced by the voltammetry curves in Figure 10G, the MSE electrode (red line) exhibited a two-fold higher baseline current and a three-fold greater signal gain than the planar electrode (black line). These results demonstrate precise detection over a broad concentration range of 0.3-200 μM. These findings highlight that strategic interface functionalization and geometric optimization are key to extending sensing ranges and developing high-performance platforms for diverse biological environments.
Levodopa
Levodopa (L-Dopa) is the metabolic precursor of dopamine and the primary clinical agent for restoring dopaminergic neurotransmission in the central nervous system[213,214]. Unlike dopamine, L-Dopa can efficiently cross the blood-brain barrier via amino acid transporters, making it the preferred therapeutic molecule for restoring dopamine levels in the brain[215]. Despite its pharmacological advantages, the therapeutic effect of L-Dopa is highly sensitive to its systemic concentration[216,217]. Progressive disease stages shorten the half-life of L-Dopa and narrow its therapeutic window. This narrowing leads to motor fluctuations and dyskinesia during overdosing, or a loss of symptom control when underdosed. As a result, precise monitoring of L-Dopa levels has become essential for optimizing treatment outcomes[217].
These clinical challenges are most evident in Parkinson’s disease, a chronic neurodegenerative disorder characterized by the loss of dopaminergic neurons in the substantia nigra[218]. While replenishing dopamine is fundamental to treatment, peripheral dopamine administration induces severe cardiovascular and gastrointestinal side effects and fails to reach the brain[219,220]. Consequently, L-Dopa has become the gold standard pharmacotherapy, and its concentration must be tightly regulated throughout long-term disease management[218,221].
Figure 11A illustrates L-Dopa metabolism and the corresponding detection principles. In vivo, L-Dopa is converted to dopamine through decarboxylation by aromatic L-amino acid decarboxylase:
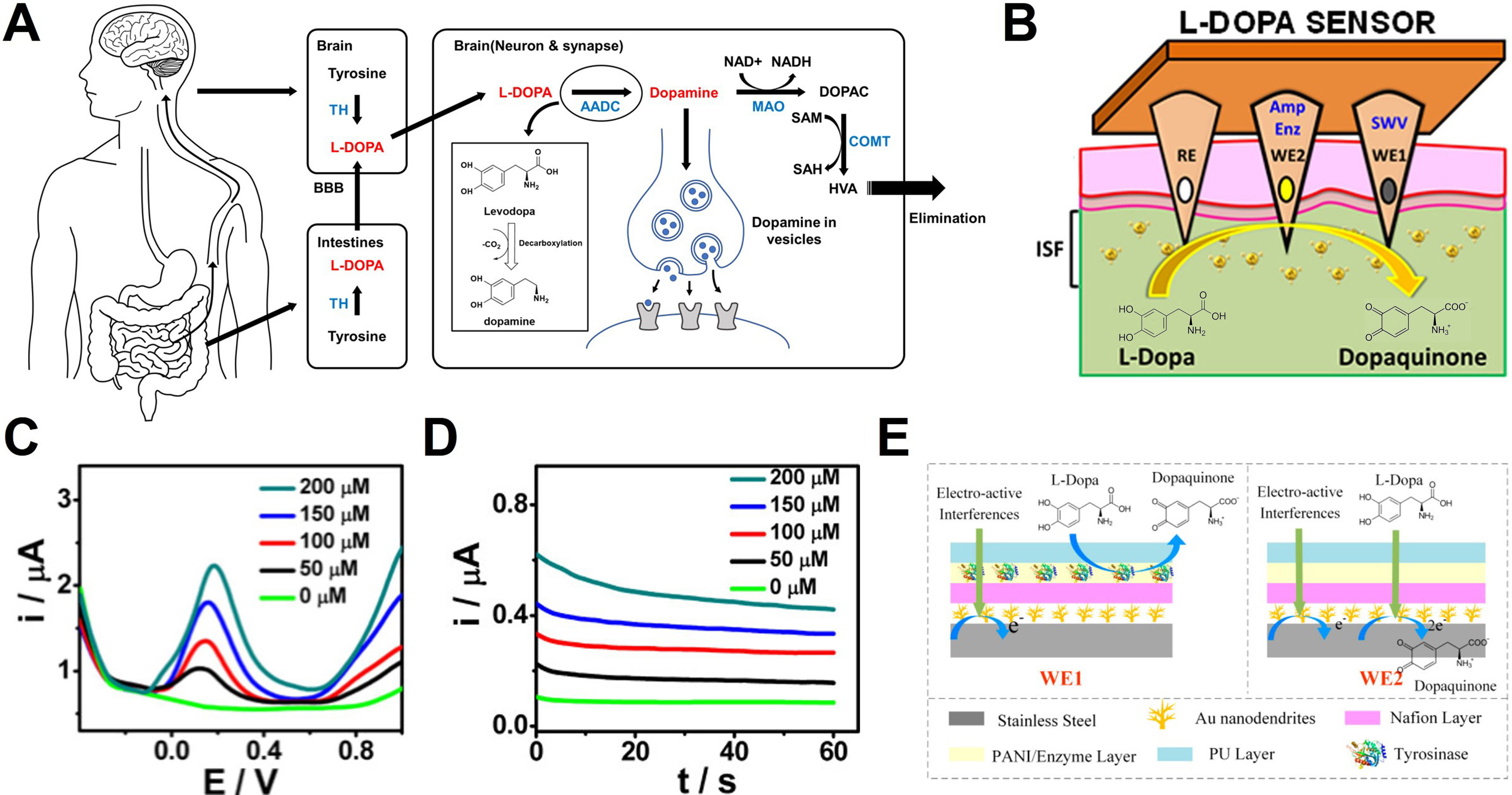
Figure 11. Dopamine modulation by L-Dopa and real-time biosensing. (A) Schematic diagram illustrating the in vivo administration route and metabolic process of L-Dopa, leading to the formation and excretion of dopamine; (B) Schematic illustration of the sensor configuration and sensing principle for L-Dopa detection. The sensor detects the electron transfer generated at the WE during the oxidation of L-Dopa to dopaquinone, utilizing a dual-electrode system for enzymatic and non-enzymatic measurements; (C) SWV and (D) chronoamperometric results corresponding to increasing L-Dopa concentrations (0-200 µM); (E) Schematic of the differential sensing mechanism and the applied sensor. WE1 selectively detects interferences by blocking the L-Dopa reaction, while WE2 detects all electro-active substances. The specific signal of L-Dopa is extracted by calculating the difference between the two signals. (B-D) Reproduced with permission Copyright 2019, ACS Sensors[222]. (E) Reproduced with permission Copyright 2022, Biosensors[223]. L-Dopa: Levodopa; WE: working electrode; SWV: square wave voltammetry; TH: tyrosine hydroxylase; L-DOPA: levodopa; BBB: blood-brain barrier; AADC: aromatic L-amino acid decarboxylase; DOPAC: 3,4-dihydroxyphenylacetic acid; MAO: monoamine oxidase; NAD+: nicotinamide adenine dinucleotide (oxidized form); NADH: nicotinamide adenine dinucleotide (reduced form); SAM: S-adenosyl-L-methionine; SAH: S-adenosyl-L-homocysteine; COMT: catechol-O-methyltransferase; HVA: homovanillic acid; PANI: polyaniline; PU: polyurethane.
In contrast, the electrochemical sensors discussed in this section quantify L-Dopa based on its oxidation to dopaquinone, during which two electrons and two protons are released:
Using this electrochemical mechanism, Goud et al. proposed an orthogonal sensing system for stable monitoring of L-Dopa in ISF[222]. The device consists of hollow MNs filled with carbon paste and simultaneously operates non-enzymatic square wave voltammetry (SWV) and enzymatic amperometry to cross-validate the measurement outputs. Figure 11B shows the structure of dual-WE system and the sensing performance in phantom gel models. The two sensing modes exhibited clear and proportional signal variations across a concentration range of 0 -200 μM [Figure 11C and D]. In SWV, the oxidation peak current increased with concentration, whereas amperometric signals displayed concentration-dependent current magnitudes, confirming the reliability of in situ L-Dopa monitoring.
To eliminate interference from high concentrations of endogenous biomolecules, Fang et al. developed a flexible differential MNA (FDMA) incorporating a differential sensing strategy[223]. Figure 11E illustrates the operational principle based on the structural differences between the reference WE1 and the sensing WE2. WE1 is coated with a tyrosinase layer that pre-consumes L-Dopa, allowing detection of only interfering species, whereas WE2 captures the total signal. Subtracting the two measurements effectively cancels interference-derived noise. This differential design achieved high linearity and sensitivity with minimal interference under various interfering conditions including serum matrix.
Tobramycin
Tobramycin is a potent aminoglycoside antibiotic primarily utilized to treat severe Gram-negative bacterial infections, including those caused by Pseudomonas aeruginosa[224,225]. Due to its narrow therapeutic index, maintaining tobramycin concentrations within a precise clinical range is critical to prevent serious adverse effects such as nephrotoxicity and ototoxicity. Consequently, there is a significant clinical demand for rapid and reliable TDM tools capable of replacing time-consuming, laboratory based diagnostics.
Wu et al. developed a device-level MN electrochemical aptamer-based (MN-EAB) sensor incorporating integrated electronics and a battery, thereby enabling continuous real-time monitoring of tobramycin in ISF. Figure 12A depicts the wearable patch architecture, the scheme for in vivo application, and histological evidence of skin penetration traces following MN insertion. Histological analysis confirms that the MNs successfully penetrate the epidermis to reach the vascularized dermis with minimal tissue disruption, ensuring rapid recovery at the insertion site. The in vivo sensing performance of the device was evaluated using SWV (Vrms = 25 mV, frequency = 80 Hz) [Figure 12B][131]. Following intravenous administration of tobramycin (20 mg/kg) in a rat model, a distinct redox peak was observed near -0.28 V (vs. Ag/AgCl)[131]. The corresponding peak current then decreased progressively over time, particularly after 50 min. This behavior indicates that the distribution, redistribution, and elimination of the drug within ISF can be tracked in real-time.
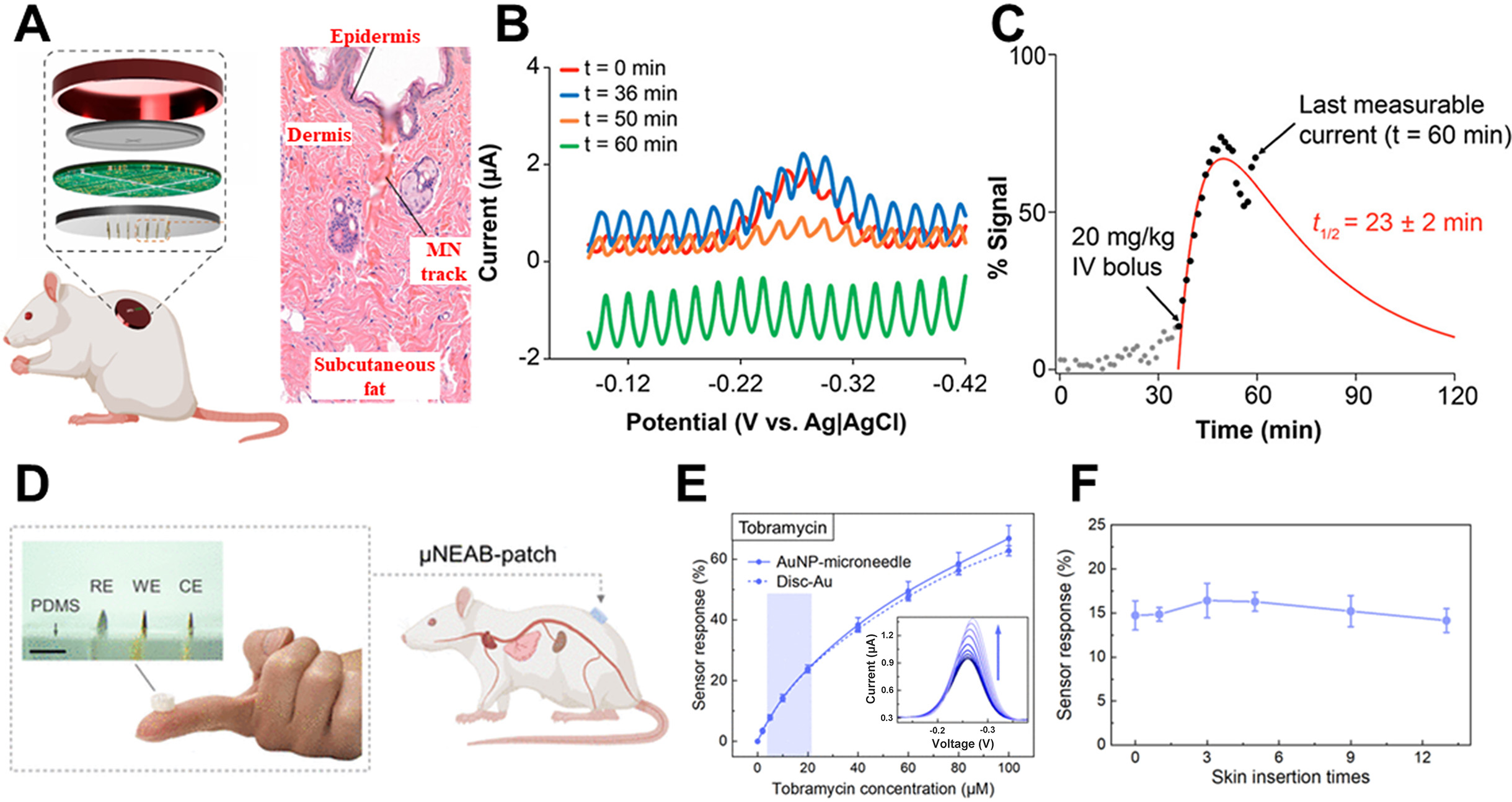
Figure 12. Electrochemical aptamer-based tobramycin sensors for minimally invasive therapeutic drug monitoring. (A) Schematic illustration of a MN patch applied to a rodent model, together with stained histological images showing penetration marks remaining in the skin after MN insertion; (B) Raw SWV recorded from rat ISF; (C) Real-time pharmacokinetic profile extracted from noisy raw voltammograms through algorithmic signal processing; (D) Schematic overview of an AuNP-coated MN-EAB patch designed for minimally invasive ISF microsampling and efficient signal transduction; (E) Tobramycin sensing response of the MN-EAB platform compared with that of a standard gold disk EAB sensor. The inset shows raw SWV acquired over a tobramycin concentration range of 0-100 µM; (F) Response of the MN-EAB sensor to 10 µM tobramycin during repeated insertion into porcine skin tissue, demonstrating the mechanical robustness of the electrochemical interface under repeated sampling conditions. (A-C) Reproduced with permission Copyright 2022, Analytical Chemistry[131]. (D-F) Reproduced with permission Copyright 2022, Science Advances[226]. MN: Microneedle; SWV: square wave voltammetry; ISF: interstitial fluid; AuNP: Au nanoparticle; EAB: electrochemical aptamer-based; PDMS: polydimethylsiloxane; RE: reference electrode; WE: working electrode; CE: counter electrode; μNEAB: microneedle-based electrochemical aptamer biosensing; Disc-Au: gold disk electrode.
To minimize noise inherent to the in vivo measurement environment, a real-time signal-processing algorithm was applied to extract a clear pharmacokinetic profile from the peak-current response [Figure 12C][131]. The processed data successfully resolved the rapid absorption phase, a short plateau region, and the subsequent elimination phase in ISF. Pharmacokinetic analysis based on a two-compartment model yielded an elimination half-life of 23 ± 2 min[131]. This value is consistent with previously reported plasma-based measurements. This supports the premise that minimally invasive ISF sampling can serve as a reliable surrogate for systemic pharmacokinetics. In summary, this study demonstrates the successful fabrication of MNA via micro-engineering and establishes the feasibility of integrating them with electrochemical aptamer-based sensors for wearable therapeutic drug monitoring.
Shuyu et al. streamlined the fabrication of MN-based electrochemical aptamer biosensors (MN-EABs) by employing a one-step Au nanoparticle (AuNP) deposition. This approach eliminates the tedious multistep procedures conventionally required for MN-EAB construction[226]. The AuNP coating provides a favorable interface for stable aptamer immobilization through thiol-based self-assembled monolayers, while also enhancing conductivity and effective surface area, both of which contribute to improved signal quality.
The performance of the fabricated sensor was evaluated in a mouse model using a tobramycin MN-EAB platform. Real-time ISF measurements and blood-ISF correlations were used to quantify in vivo tobramycin levels for dosage assessment [Figure 12D][226]. To compare the analytical performance of the AuNP-coated MN-EAB sensor with that of a conventional Au disk electrode, both platforms were functionalized with an anti-tobramycin aptamer and characterized by voltammetric analysis [Figure 12E][226]. The two devices exhibited comparable sensitivities over a tobramycin concentration range of 2-100 μM, indicating that the AuNP-coated MN achieved signal stability and sensitivity equivalent to those of the standard Au disk electrode[226].
The mechanical and electrochemical stability of the AuNP-coated MN-EAB sensor was further examined through repeated insertion into porcine skin samples containing 10 μM tobramycin. As shown in Figure 12F, the sensor maintained an approximately 15% signal response after repeated insertions, indicating that the AuNP and aptamer layers remained stably anchored to the MN surface[226]. This result suggests improved resistance to insertion-induced damage compared with conventional deposited metal films.
Finally, the pharmacokinetic behavior of tobramycin in ISF was monitored in a rat model. Following intravenous administration of tobramycin at doses of 10-30 mg/kg, both the maximum sensor response and the area under the ISF response curve over 80 min after dosing increased in a dose-dependent manner. These findings demonstrate that the AuNP-based MN-EAB platform enables reliable real-time monitoring of in vivo tobramycin dynamics. They also suggest that this platform may provide a practical basis for dosage-guided therapeutic monitoring.
CLOSED-LOOP SYSTEM
This section introduces a closed-loop system capable of simultaneously diagnosing physiological abnormalities and administering therapeutic treatments. Wei et al. proposed an integrated platform based on MNA that monitors in vivo glucose levels in real-time and actively delivers insulin in response. Figure 13A shows the system attached to a diabetic rat model, a setup designed to verify biocompatibility and validate the insulin delivery function in a living organism[227]. The platform is engineered to interface with a smartphone via Bluetooth, enabling real-time wireless data transmission and remote control.
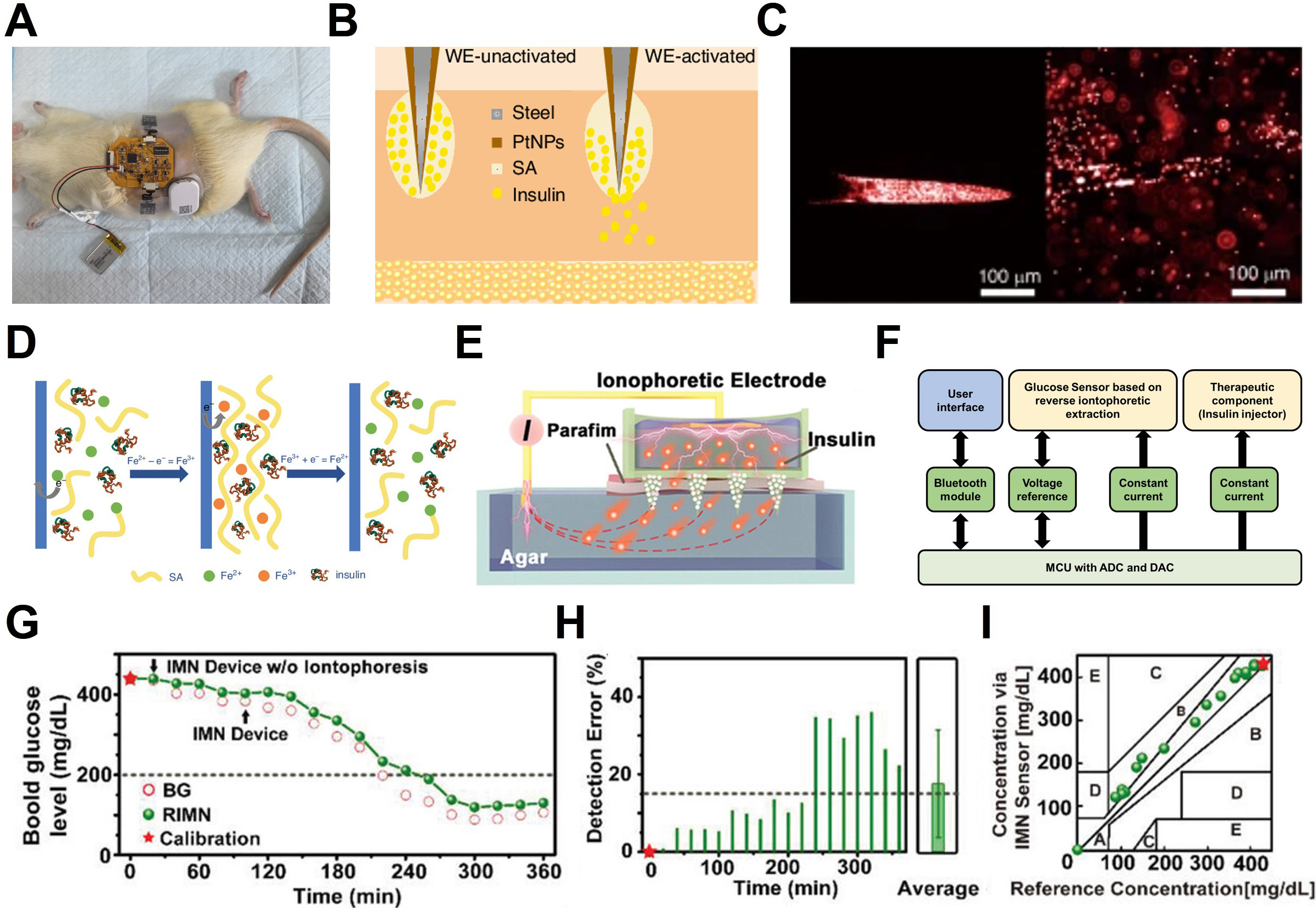
Figure 13. Closed-loop glucose monitoring system for diabetes management. (A) In vivo evaluation of the integrated wearable closed-loop system (IWCS) on a diabetic rat; (B) Schematic of the electrochemically triggered insulin release electrode; (C) Fluorescence images visualizing insulin release before (left) and after (right) electrical triggering; (D) Mechanism of the redox-responsive Fe3+/Fe2+ hydrogel sol-gel transition; (E) Schematic of iontophoretic insulin delivery in an in vitro agar model; (F) System block diagram for closed-loop glucose monitoring; (G) Comparison of real-time glucose monitoring between the RIMN sensor and standard blood glucose (BG) measurements; (H) Statistical analysis of detection error; (I) Clarke’s error grid analysis confirming clinical accuracy. (A-D) Reproduced with permission Copyright 2025, Microsystems & Nanoengineering[227]. (E, G-I) Reproduced with permission Copyright 2021, Advanced Science[77]. RIMN: Reverse iontophoresis microneedle; WE: working electrode; NPs: nanoparticles; SA: sodium alginate; MCU: microprogrammed control unit; ADC: analog-to-digital converter; DAC: digital-to-analog converter; IMN: iontophoretic microneedle.
The glucose sensing mechanism follows the first-generation enzymatic principle described in Chapter 2. Figure 13B presents a cross-sectional view of the insulin release module. To visually verify the release behavior, the researchers used a hydrogel precursor solution containing fluorescent microspheres. As shown in Figure 13C, fluorescence microscopy confirmed that the hydrogel dissociates and releases the encapsulated content upon the application of an electrical stimulus.
Figure 13D illustrates the core mechanism of this active release control. Sodium alginate (SA) forms a rigid hydrogel by strongly crosslinking with Fe3+, whereas it exhibits low binding affinity for Fe2+, preventing stable gel formation. The system operates on this reversible Fe3+/Fe2+ redox transition. Applying an oxidation voltage oxidizes Fe2+ to Fe3+ to encapsulate insulin via gelation. Conversely, applying a reduction potential (e.g., -1.5 V) reduces Fe3+ back to Fe2+, triggering a gel-sol transition that dissolves the network and releases the drug[227]. Unlike passive diffusion, this electrochemical actuation enables precise “on-demand” insulin administration based on real-time glucose monitoring.
Despite these advantages, the diagnosis-and-treatment system shown in Figure 13A-C relies on spontaneous drug release triggered by a gel-sol phase transition, which limits precise dose control and immediate feedback. In addition, the system requires users to manually activate a switch through a smartphone application upon receiving a hyperglycemia alert. This prevents it from functioning as a fully autonomous closed-loop platform. In contrast, the system illustrated in Figure 13E integrates a microprogrammed control unit (MCU) that autonomously initiates electrophoretic insulin release when hyperglycemia is detected[77]. This platform comprises three primary components: a sensing module, a computation unit, and an infusion module. Analog signals generated by the sensor are transmitted to the MCU and digitized via an analog-to-digital converter (ADC). The processed data are then relayed through a wireless transceiver. Upon detection of a hyperglycemic event, the MCU applies a direct current (DC) signal to the insulin infusion unit, thereby triggering controlled electrophoretic insulin release. Unlike passive methods relying solely on concentration gradients, the application of a specific current (e.g., 0.5 mA) generates an electric field that actively repels charged insulin molecules [Figure 13F]. This mechanism significantly accelerates their transport through mesoporous MNs into the subcutaneous tissue. This active transport effectively overcomes the lag time and low permeability inherent to passive transdermal systems, enabling precise, on-demand dosage control.
The efficacy of this rapid delivery logic was validated in a diabetic rat model. Upon detecting hyperglycemia via the reverse iontophoresis microneedle (RIMN) sensor, the system triggered the iontophoretic device. Figure 13G demonstrates that blood glucose levels decreased immediately after activation, reaching the normoglycemic range (< 200 mg/dL) within approximately 1.5 h. Furthermore, sensing accuracy remained stable despite the simultaneous operation of the delivery module. Figure 13H indicates an average detection error of 17.5% ± 13.9%. Finally, Clarke Error Grid Analysis confirmed clinical acceptability, with 73.7% of data points falling within Zone A [Figure 13I]. By successfully miniaturizing this “sense-act-treat” feedback loop, the IWCS offers a promising, compact alternative to bulky pump-sensor setups.
CHALLENGES AND OUTLOOK FOR CLINICAL TRANSLATION
Electrochemical biosensing is rapidly advancing toward multimodal biochemical monitoring. However, transitioning these technologies into routine clinical practice requires addressing several critical translational challenges. First, most emerging sensors have not yet achieved the analytical rigor necessary for clinical deployment. Robust standardized calibration requires a reproducible relationship between sensor output and true analyte concentration. This relationship must remain stable across individuals, device lots, and varying environmental conditions throughout the sensor’s operational lifetime. The extensive clinical history of glucose monitors has established a high benchmark for calibration. However, this level of reliability remains unmatched by metabolic, hormonal, or pharmacological sensing platforms, especially under fluctuating physiological conditions. Equally essential is long-term stability, with reliable operation spanning days for acute monitoring and weeks to months for wearable tracking. Ensuring such durability necessitates overcoming protein fouling, enzymatic degradation, and electrode corrosion. In particular, mitigating fibrotic encapsulation and electrode corrosion remains a critical frontier for material science innovation[228]. Also, meeting regulatory-grade expectations requires compliance with FDA and ISO standards, such as the 15/15 accuracy criteria and rigorous interference testing. However, these frameworks are not yet broadly established for non-glucose analytes.
Second, the integration of biosensing with adaptive therapeutic decision-making remains at an early stage. This includes applications such as drug dosing, metabolic stabilization, and closed-loop automation, all of which require several enabling technologies. Reliable closed-loop control relies on programmable drug-delivery platforms, such as microfluidic pumps and electroresponsive polymers. Clinical deployment will further require expansive, multimodal training datasets that mirror complex clinical decision-making. These datasets must integrate real-time sensor readouts with clinical laboratory values, medication histories, symptom reports, and physiological covariates to ensure safe and interpretable automation. In parallel, fault-tolerant architectures are essential to compensate for sensor drift and biological variability, thereby supporting personalized disease management. Collectively, these elements, when integrated through AI-driven frameworks, could enable autonomous biochemical regulation and more personalized therapeutic control[229].
Finally, achieving reliable multimodal monitoring requires architectures capable of overcoming the large concentration disparity between millimolar metabolites and nanomolar signaling molecules[24,78]. This challenge can be mitigated by employing diffusion-regulating membranes (e.g., Nafion, PU) to prevent enzyme overload in high-concentration sensors[230]. Simultaneously, the use of nanostructured interfaces or 3D microstructured electrodes can maximize the effective surface area for low-concentration targets, thereby enhancing the signal-to-noise ratio and sensitivity[231]. Furthermore, combining differential sensing strategies with conductive antifouling layers will be essential for maintaining the analytical precision required for clinically actionable monitoring[77,124].
CONCLUSION
Electrochemical biosensing is evolving beyond traditional single-analyte detection toward comprehensive platforms for monitoring metabolites, hormones, and therapeutic agents across microsampled blood and minimally invasive ISF. This review has established a practical framework for selecting device architectures and measurement strategies tailored to specific target biomarkers and their physiological contexts. Recent advances in sampling-aware sensor design, surface engineering, and wearable electronics have laid the foundation for quantitative biochemical monitoring with unprecedented temporal resolution. A central insight from the discussed literature is that biofluid selection is not merely a matter of practical convenience. Rather, it is a fundamental design variable that determines target accessibility, calibration requirements, and clinical interpretability. Blood microsampling remains advantageous for analytes enriched in systemic circulation or poorly partitioned into ISF, particularly high-molecular-weight targets. Conversely, ISF offers an optimal matrix for minimally invasive, continuous monitoring. Consequently, successful platform translation necessitates the rigorous alignment of the target analyte, its physiological distribution, and the intended clinical application with the appropriate sampling strategy and detection modality. Compared to optical and mass spectrometric methodologies, electrochemical biosensors are distinctly positioned for integration into portable, wearable, and user-operated systems. By directly transducing biochemical events into electrical signals, electrochemical platforms bypass the need for bulky optical components or high-vacuum environments, enabling structural simplicity, extreme miniaturization, and superior power efficiency. Such attributes are critical for long-term continuous monitoring, therapeutic drug management, and POC decision support. The convergence of flexible electrode arrays, MN interfaces, highly integrated low-power electronics, and wireless data transmission is rapidly accelerating the practical deployment of these biosensing systems. Nevertheless, substantial barriers to clinical translation persist. Key challenges include ensuring long-term sensor stability and antifouling performance in complex biofluids, as well as achieving reproducible and scalable manufacturing. Additional priorities include realizing multiplexed detection across broad dynamic ranges, developing standardized in vivo calibration protocols, and conducting rigorous validation against established clinical outcomes. Overcoming these translational hurdles is imperative for transitioning electrochemical biosensors from promising experimental concepts to validated clinical tools. Ultimately, the synergistic integration of multiplexed quantitative sensing with closed-loop therapeutic interventions holds the potential to elevate electrochemical biosensing from a passive monitoring modality to an active, foundational component of personalized precision medicine.
DECLARATIONS
Authors’ contributions
Contributed to the investigation, visualization and writing: Yoon, S. J.; Park, J. T.; Yun, J. M.
Contributed to conceptualization, writing, supervision: Lee, Y. K.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (version 5.4, released 2026-03-06) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by the Fund of Biomedical Research Institute of Jeonbuk National University Hospital, and the Ministry of Trade, Industry & Energy (MOTIE) and the Ministry of Science and ICT (MSIT) of the Korea government (Grant Nos. RS-2025-25441263, RS-2025-02306270).
Conflicts of interest
Not applicable.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Heller, A.; Feldman, B. Electrochemical glucose sensors and their applications in diabetes management. Chem. Rev. 2008, 108, 2482-505.
2. Clark, L. C.; Lyons, C. Electrode systems for continuous monitoring in cardiovascular surgery. Ann. N. Y. Acad. Sci. 2006, 102, 29-45.
3. Wang, J. Electrochemical biosensors: towards point-of-care cancer diagnostics. Biosens. Bioelectron. 2006, 21, 1887-92.
4. Wu, J.; Liu, H.; Chen, W.; Ma, B.; Ju, H. Device integration of electrochemical biosensors. Nat. Rev. Bioeng. 2023, 1, 346-60.
5. Zhang, H.; Sun, Z.; Sun, K.; et al. Electrochemical impedance spectroscopy-based biosensors for label-free detection of pathogens. Biosensors 2025, 15, 443.
6. Jung, H. H.; Lee, H.; Yea, J.; Jang, K. Wearable electrochemical sensors for real-time monitoring in diabetes mellitus and associated complications. Soft. Sci. 2024, 4, 15.
7. Pour, S. R. S.; Calabria, D.; Emamiamin, A.; et al. Electrochemical vs. optical biosensors for point-of-care applications: a critical review. Chemosensors 2023, 11, 546.
8. Zhang, Y.; Zhou, N. Electrochemical biosensors based on micro-fabricated devices for point-of-care testing: a review. Electroanalysis 2021, 34, 168-83.
9. Umapathi, R.; Raju, C. V.; Safarkhani, M.; et al. Versatility of MXene based materials for the electrochemical detection of phenolic contaminants. Coord. Chem. Rev. 2025, 525, 216305.
10. Umapathi, R.; Ghoreishian, S. M.; Sonwal, S.; Rani, G. M.; Huh, Y. S. Portable electrochemical sensing methodologies for on-site detection of pesticide residues in fruits and vegetables. Coord. Chem. Rev. 2022, 453, 214305.
11. Bandodkar, A. J.; Jeerapan, I.; Wang, J. Wearable chemical sensors: present challenges and future prospects. ACS. Sens. 2016, 1, 464-82.
12. Falk, M.; Psotta, C.; Cirovic, S.; Shleev, S. Non-invasive electrochemical biosensors operating in human physiological fluids. Sensors 2020, 20, 6352.
13. Shen, Y.; Liu, C.; He, H.; et al. Recent advances in wearable biosensors for non-invasive detection of human lactate. Biosensors 2022, 12, 1164.
14. Lad, U.; Khokhar, S.; Kale, G. M. Electrochemical creatinine biosensors. Anal. Chem. 2008, 80, 7910-7.
15. Yeasmin, S.; Cheng, L. Emerging trends in functional molecularly imprinted polymers for electrochemical detection of biomarkers. Biomicrofluidics 2024, 18, 031503.
16. Li, Y.; Luo, L.; Kong, Y.; et al. Recent advances in molecularly imprinted polymer-based electrochemical sensors. Biosens. Bioelectron. 2024, 249, 116018.
17. He, R.; Chen, L.; Chu, P.; Gao, P.; Wang, J. Recent advances in nonenzymatic electrochemical biosensors for sports biomarkers: focusing on antibodies, aptamers and molecularly imprinted polymers. Anal. Methods. 2024, 16, 6079-97.
18. Wang, W.; He, Y.; He, S.; et al. A brief review of aptamer-based biosensors in recent years. Biosensors 2025, 15, 120.
19. Kim, Y.; Seo, M.; Baek, S. Ion-selective electrode-based sensors from the macro- to the nanoscale. Sensor. Actuator. Rep. 2025, 9, 100258.
20. Cha, S.; Choi, M. Y.; Kim, M. J.; Sim, S. B.; Haizan, I.; Choi, J. Electrochemical microneedles for real-time monitoring in interstitial fluid: emerging technologies and future directions. Biosensors 2025, 15, 380.
21. Yuan, X.; Ouaskioud, O.; Yin, X.; et al. Epidermal wearable biosensors for the continuous monitoring of biomarkers of chronic disease in interstitial fluid. Micromachines 2023, 14, 1452.
22. Pereira, R. L.; Vinayakumar, K. B.; Sillankorva, S. Polymeric microneedles for health care monitoring: an emerging trend. ACS. Sens. 2024, 9, 2294-309.
23. Haider, K.; Dalton, C. Recent developments in microneedle biosensors for biomedical and agricultural applications. Micromachines 2025, 16, 929.
24. Tehrani, F.; Teymourian, H.; Wuerstle, B.; et al. An integrated wearable microneedle array for the continuous monitoring of multiple biomarkers in interstitial fluid. Nat. Biomed. Eng. 2022, 6, 1214-24.
25. Dervisevic, M.; Esser, L.; Chen, Y.; Alba, M.; Prieto-simon, B.; Voelcker, N. H. High-density microneedle array-based wearable electrochemical biosensor for detection of insulin in interstitial fluid. Biosens. Bioelectron. 2025, 271, 116995.
26. Duan, H.; Peng, S.; He, S.; et al. Wearable electrochemical biosensors for advanced healthcare monitoring. Adv. Sci. 2024, 12, 2411433.
27. Dong, Y.; Mao, S.; Chen, S.; Ma, J.; Jaffrezic-renault, N.; Guo, Z. Opportunities and challenges of microneedle electrochemical sensors for interstitial fluid detection. TrAC - Trends Anal.. Chem. 2024, 180, 117891.
28. Yin, S.; Yu, Z.; Song, N.; et al. A long lifetime and highly sensitive wearable microneedle sensor for the continuous real-time monitoring of glucose in interstitial fluid. Biosens. Bioelectron. 2024, 244, 115822.
29. Byrne, B.; Stack, E.; Gilmartin, N.; O’kennedy, R. Antibody-based sensors: principles, problems and potential for detection of pathogens and associated toxins. Sensors 2009, 9, 4407-45.
30. Aramburo, A.; Todd, J.; George, E. C.; et al. Lactate clearance as a prognostic marker of mortality in severely ill febrile children in East Africa. BMC. Med. 2018, 16, 37.
31. Kruse, O.; Grunnet, N.; Barfod, C. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scand. J. Trauma. Resusc. Emerg. Med. 2011, 19, 74.
32. Ming, D. K.; Jangam, S.; Gowers, S. A. N.; et al. Real-time continuous measurement of lactate through a minimally invasive microneedle patch: a phase I clinical study. BMJ. Innov. 2022, 8, 87-94.
33. Leung, H. M. C.; Forlenza, G. P.; Prioleau, T. O.; Zhou, X. Noninvasive glucose sensing in vivo. Sensors 2023, 23, 7057.
34. Wang, Y.; Vaddiraju, S.; Gu, B.; Papadimitrakopoulos, F.; Burgess, D. J. Foreign body reaction to implantable biosensors: effects of tissue trauma and implant size. J. Diabetes. Sci. Technol. 2015, 9, 966-77.
35. Chen, Y.; He, Z.; Wu, Y.; et al. A wearable molecularly imprinted electrochemical sensor for cortisol stable monitoring in sweat. Biosensors 2025, 15, 194.
36. Singh, N. K.; Chung, S.; Sveiven, M.; Hall, D. A. Cortisol detection in undiluted human serum using a sensitive electrochemical structure-switching aptamer over an antifouling nanocomposite layer. ACS. Omega. 2021, 6, 27888-97.
37. Wu, Z.; Qiao, Z.; Chen, S.; et al. Interstitial fluid-based wearable biosensors for minimally invasive healthcare and biomedical applications. Commun. Mater. 2024, 5, 33.
38. Kim, G.; Ahn, H.; Chaj Ulloa, J.; Gao, W. Microneedle sensors for dermal interstitial fluid analysis. Med-X 2024, 2, 15.
39. Sprunger, Y.; Longo, J.; Saeidi, A.; Ionescu, A. M. Bridging blood and skin: biomarker profiling in dermal interstitial fluid (dISF) for minimally invasive diagnostics. Biosensors 2025, 15, 301.
40. Himawan, A.; Vora, L. K.; Permana, A. D.; et al. Where microneedle meets biomarkers: futuristic application for diagnosing and monitoring localized external organ diseases. Adv. Healthc. Mater. 2022, 12, 2202066.
41. Friedel, M.; Thompson, I. A. P.; Kasting, G.; et al. Opportunities and challenges in the diagnostic utility of dermal interstitial fluid. Nat. Biomed. Eng. 2023, 7, 1541-55.
42. Levick, J. R.; Michel, C. C. Microvascular fluid exchange and the revised Starling principle. Cardiovasc. Res. 2010, 87, 198-210.
43. Heikenfeld, J.; Jajack, A.; Feldman, B.; et al. Accessing analytes in biofluids for peripheral biochemical monitoring. Nat. Biotechnol. 2019, 37, 407-19.
44. Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenesis. Res. 2010, 2, 14.
45. Frank, P. G.; Pavlides, S.; Lisanti, M. P. Caveolae and transcytosis in endothelial cells: role in atherosclerosis. Cell. Tissue. Res. 2008, 335, 41-7.
46. Basu, A.; Dube, S.; Slama, M.; et al. Time lag of glucose from intravascular to interstitial compartment in humans. Diabetes 2013, 62, 4083-7.
47. Thennadil, S. N.; Rennert, J. L.; Wenzel, B. J.; Hazen, K. H.; Ruchti, T. L.; Block, M. B. Comparison of glucose concentration in interstitial fluid, and capillary and venous blood during rapid changes in blood glucose levels. Diabetes. Technol. Ther. 2001, 3, 357-65.
48. Keenan, D. B.; Mastrototaro, J. J.; Voskanyan, G.; Steil, G. M. Delays in minimally invasive continuous glucose monitoring devices: a review of current technology. J. Diabetes. Sci. Technol. 2009, 3, 1207-14.
49. Kolluru, C.; Williams, M.; Yeh, J. S.; Noel, R. K.; Knaack, J.; Prausnitz, M. R. Monitoring drug pharmacokinetics and immunologic biomarkers in dermal interstitial fluid using a microneedle patch. Biomed. Microdevices. 2019, 21, 14.
50. Samant, P. P.; Niedzwiecki, M. M.; Raviele, N.; et al. Sampling interstitial fluid from human skin using a microneedle patch. Sci. Transl. Med. 2020, 12, eaaw0285.
51. Friedel, M.; Werbovetz, B.; Drexelius, A.; et al. Continuous molecular monitoring of human dermal interstitial fluid with microneedle-enabled electrochemical aptamer sensors. Lab. Chip. 2023, 23, 3289-99.
52. Geyer, P. E.; Kulak, N. A.; Pichler, G.; Holdt, L. M.; Teupser, D.; Mann, M. Plasma proteome profiling to assess human health and disease. Cell. Systems. 2016, 2, 185-95.
53. Anderson, N. L.; Anderson, N. G. The human plasma proteome. Mol. Cell. Proteomics. 2002, 1, 845-67.
54. Corrie, S. R.; Coffey, J. W.; Islam, J.; Markey, K. A.; Kendall, M. A. F. Blood, sweat, and tears: developing clinically relevant protein biosensors for integrated body fluid analysis. Analyst 2015, 140, 4350-64.
55. Jairam, R. K.; Franz, M.; Hanke, N.; Kuepfer, L. Physiologically based pharmacokinetic models for systemic disposition of protein therapeutics in rabbits. Front. Pharmacol. 2024, 15, 1427325.
56. Chen, X.; Crimmins, E.; Hu, P. P.; et al. Venous blood-based biomarkers in the china health and retirement longitudinal study: rationale, design, and results from the 2015 wave. Am. J. Epidemiol. 2019, 188, 1871-7.
57. Grande, G.; Valletta, M.; Rizzuto, D.; et al. Blood-based biomarkers of Alzheimer’s disease and incident dementia in the community. Nat. Med. 2025, 31, 2027-35.
58. Sly, B.; Taylor, J. Blood glucose monitoring devices: current considerations. Aust. Prescr. 2023, 46, 54-9.
59. Rebel, A.; Rice, M. A.; Fahy, B. G. The accuracy of point-of-care glucose measurements. J. Diabetes. Sci. Technol. 2012, 6, 396-411.
60. Chan, J. C.; Wong, R. Y.; Cheung, C.; et al. Accuracy, precision and user-acceptability of self blood glucose monitoring machines. Diabetes. Res. Clin. Pract. 1997, 36, 91-104.
61. Sharif-nia, H.; Mokhtari, H.; Osborne, J. W.; Shafaei, S.; Soltanzade, M. Evaluation of the accuracy, precision, and agreement of a glucometer compared to the standard laboratory test in diabetic and non-diabetic patients. Sci. Rep. 2025, 15, 44517.
62. Hoffman, M. S. F.; Mckeage, J. W.; Xu, J.; Ruddy, B. P.; Nielsen, P. M. F.; Taberner, A. J. Minimally invasive capillary blood sampling methods. Expert. Rev. Med. Devices. 2023, 20, 5-16.
63. Fruhstorfer, H.; Schmelzeisen-redeker, G.; Weiss, T. Capillary blood sampling: relation between lancet diameter, lancing pain and blood volume. Eur. J. Pain. 2012, 3, 283-6.
64. Oliveira, C.; Teixeira, J. A.; Oliveira, N.; Ferreira, S.; Botelho, C. M. Microneedles’ device: design, fabrication, and applications. Macromol 2024, 4, 320-55.
65. Kim, J.; Park, S.; Nam, G.; Choi, Y.; Woo, S.; Yoon, S. Bioinspired microneedle insertion for deep and precise skin penetration with low force: why the application of mechanophysical stimuli should be considered. J. Mech. Behav. Biomed. Mater. 2018, 78, 480-90.
66. Zahra Jawad, S. E.; Hussain, D.; Najam-ul-haq, M.; Fatima, B. Electrochemical sensing of human hormones. TrAC. -. Trends. Anal. Chem. 2024, 181, 117993.
67. Mikuła, E. Recent advancements in electrochemical biosensors for Alzheimer’s disease biomarkers detection. Curr. Med. Chem. 2021, 28, 4049-73.
68. Gunasekaran, A. K.; Nesakumar, N.; Gunasekaran, B. M.; Kulandaisamy, A. J.; Balaguru Rayappan, J. B. Highly sensitive non-enzymatic electrochemical sensor for uric acid detection using copper oxide nanopebbles-modified glassy carbon electrode. Appl. Surf. Sci. 2025, 697, 162956.
69. Wang, L.; Chang, S.; Chen, C.; Liu, J. Metal-organic frameworks for electrochemical glucose sensors: progress and challenges. Coord. Chem. Rev. 2025, 543, 216907.
70. Kim, J.; Campbell, A. S.; Wang, J. Wearable non-invasive epidermal glucose sensors: a review. Talanta 2018, 177, 163-70.
71. Smoller, B. R.; Roe, J. N. Bloodless glucose measurements. Crit. Rev. Ther. Drug. Carrier. Syst. 1998, 15, 43.
72. Mcallister, D. V.; Wang, P. M.; Davis, S. P.; et al. Microfabricated needles for transdermal delivery of macromolecules and nanoparticles: fabrication methods and transport studies. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13755-60.
73. Hu, Y.; Chatzilakou, E.; Pan, Z.; Traverso, G.; Yetisen, A. K. Microneedle sensors for point-of-care diagnostics. Adv. Sci. 2024, 11, 2306560.
74. Corrie, S. R.; Fernando, G. J. P.; Crichton, M. L.; Brunck, M. E. G.; Anderson, C. D.; Kendall, M. A. F. Surface-modified microprojection arrays for intradermal biomarker capture, with low non-specific protein binding. Lab. Chip. 2010, 10, 2655.
75. Windmiller, J. R.; Valdés-ramírez, G.; Zhou, N.; et al. Bicomponent microneedle array biosensor for minimally-invasive glutamate monitoring. Electroanalysis 2011, 23, 2302-9.
76. Dardano, P.; Battisti, M.; Rea, I.; et al. Polymeric microneedle arrays: versatile tools for an innovative approach to drug administration. Adv. Ther. 2019, 2, 1900036.
77. Li, X.; Huang, X.; Mo, J.; et al. A fully integrated closed-loop system based on mesoporous microneedles-iontophoresis for diabetes treatment. Adv. Sci. 2021, 8, 2100827.
78. Kusama, S.; Sato, K.; Matsui, Y.; et al. Transdermal electroosmotic flow generated by a porous microneedle array patch. Nat. Commun. 2021, 12, 658.
79. Donnelly, R. F.; Singh, T. R. R.; Garland, M. J.; et al. Hydrogel-forming microneedle arrays for enhanced transdermal drug delivery. Adv. Funct. Mater. 2012, 22, 4879-90.
80. Mandal, A.; Boopathy, A. V.; Lam, L. K. W.; et al. Cell and fluid sampling microneedle patches for monitoring skin-resident immunity. Sci. Transl. Med. 2018, 10, eaar2227.
81. Ghavaminejad, P.; Ghavaminejad, A.; Zheng, H.; Dhingra, K.; Samarikhalaj, M.; Poudineh, M. A. Conductive hydrogel microneedle-based assay integrating PEDOT:PSS and Ag-Pt nanoparticles for real-time, enzyme-less, and electrochemical sensing of glucose. Adv. Healthc. Mater. 2022, 12, 2202362.
82. Filho, R. R.; Rocha, L. L.; Corrêa, T. D.; Pessoa, C. M. S.; Colombo, G.; Assuncao, M. S. C. Blood lactate levels cutoff and mortality prediction in sepsis - time for a reappraisal? A retrospective cohort study. Shock 2016, 46, 480-5.
83. Djassemi, O.; Chang, A.; Mcguire, W. C.; et al. Clinical evaluation of microneedle biosensors for continuous lactate monitoring in critically Ill patients. ACS. Sens. 2026, 11, 1413-24.
84. Veronica, A.; Li, Y.; Li, Y.; Hsing, I.; Nyein, H. Y. Y. Dermal-fluid-enabled detection platforms for non-invasive ambulatory monitoring. Sens. Diagn. 2023, 2, 1335-59.
85. Wang, H.; Dong, Q.; Zhao, P.; Yang, G.; Yan, Q.; Yang, Y. Iontophoresis-enhanced microneedles for interstitial fluid detection and transdermal drug delivery. J. Drug. Deliv. Sci. Technol. 2026, 119, 108158.
86. Han, S.; Yamamoto, S.; Jung, C.; Jin, D. Y.; Lee, T.; Kim, J. Wearable sensors for monitoring chronic kidney disease. Commun. Mater. 2024, 5, 153.
87. Adelaars, S.; Konings, C. J.; Cox, L.; et al. The correlation of urea and creatinine concentrations in sweat and saliva with plasma during hemodialysis: an observational cohort study. Clin. Chem. Lab. Med. 2024, 62, 1118-25.
88. Rakesh Kumar, R.; Shaikh, M. O.; Chuang, C. A review of recent advances in non-enzymatic electrochemical creatinine biosensing. Anal. Chim. Acta. 2021, 1183, 338748.
89. Kayashima, S.; Arai, T.; Kikuchi, M.; et al. Suction effusion fluid from skin and constituent analysis: new candidate for interstitial fluid. Am. J. Physiol. Circ. Physiol. 1992, 263, H1623-7.
90. Saltiel, A. R.; Kahn, C. R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799-806.
91. Li, X.; Yang, Y.; Zhang, B.; et al. Lactate metabolism in human health and disease. Signal. Transduct. Target. Ther. 2022, 7, 305.
92. Jadhav, R. B.; Patil, T.; Tiwari, A. P. Trends in sensing of creatinine by electrochemical and optical biosensors. Appl. Surf. Sci. Adv. 2024, 19, 100567.
93. Matsumoto, S.; Häberle, J.; Kido, J.; Mitsubuchi, H.; Endo, F.; Nakamura, K. Urea cycle disorders - update. J. Hum. Genet. 2019, 64, 833-47.
94. Gao, W.; Emaminejad, S.; Nyein, H. Y. Y.; et al. Fully integrated wearable sensor arrays for multiplexed in situ perspiration analysis. Nature 2016, 529, 509-14.
95. Yue Jing, L.; Fan, Y.; Zhi Chen, B.; et al. An aptamer-integrated conductive microneedle biosensor for real-time transdermal cortisol monitoring. Chem. Eng. J. 2024, 502, 157488.
96. Trusso Sfrazzetto, G.; Santonocito, R. Nanomaterials for cortisol sensing. Nanomaterials 2022, 12, 3790.
97. Venugopal, M.; Arya, S. K.; Chornokur, G.; Bhansali, S. A realtime and continuous assessment of cortisol in ISF using electrochemical impedance spectroscopy. Sens. Actuat. A. Phys. 2011, 172, 154-60.
98. Holsboer, F.; Ising, M. Stress hormone regulation: biological role and translation into therapy. Annu. Rev. Psychol. 2010, 61, 81-109.
99. Jansson, P. E.; Fowelin, J. P.; Von Schenck, H. P.; Smith, U. P.; Lönnroth, P. N. Measurement by microdialysis of the insulin concentration in subcutaneous interstitial fluid: importance of the endothelial barrier for insulin. Diabetes 1993, 42, 1469-73.
100. Pretty, C. G.; Le Compte, A.; Penning, S.; et al. Interstitial insulin kinetic parameters for a 2-compartment insulin model with saturable clearance. Comput. Methods. Programs. Biomed. 2014, 114, e39-45.
101. Sjöstrand, M.; Holmäng, A.; Lönnroth, P. Measurement of interstitial insulin in human muscle. Am. J. Physiol. -. Endocrinol. Metab. 1999, 276, E151-4.
102. Stegemann, J.; Augustin, M. N.; Ackermann, J.; et al. Levodopa sensing with a nanosensor array via a low-cost near infrared readout. Anal. Chem. 2025, 97, 13655-62.
103. Liu, Y.; Yu, Q.; Luo, X.; Yang, L.; Cui, Y. Continuous monitoring of diabetes with an integrated microneedle biosensing device through 3D printing. Microsyst. Nanoeng. 2021, 7, 75.
104. Wang, Y.; Liu, H.; Yang, X.; et al. A responsive hydrogel-based microneedle system for minimally invasive glucose monitoring. Smart. Mater. Med. 2023, 4, 69-77.
105. Misia, G.; Evangelisti, C.; Merino, J. P.; et al. Design and optimization of an electrochemical sensor based on carbon nanotubes for the reliable voltammetric detection of serotonin in complex biological fluids. Carbon 2024, 229, 119550.
106. Liu, Y.; Li, W.; Lu, Z.; et al. A microneedle-based SERS sensor for simultaneous detection of pH and uric acid in interstitial fluid. Microchem. J. 2026, 222, 117164.
107. Ju, J.; Hsieh, C.; Tian, Y.; et al. Surface enhanced raman spectroscopy based biosensor with a microneedle array for minimally invasive in vivo glucose measurements. ACS. Sens. 2020, 5, 1777-85.
108. Zhu, D. D.; Zheng, L. W.; Duong, P. K.; et al. Colorimetric microneedle patches for multiplexed transdermal detection of metabolites. Biosens. Bioelectron. 2022, 212, 114412.
109. He, R.; Niu, Y.; Li, Z.; et al. A hydrogel microneedle patch for point-of-care testing based on skin interstitial fluid. Adv. Healthc. Mater. 2020, 9, 1901201.
110. He, R.; Liu, H.; Fang, T.; et al. A colorimetric dermal tattoo biosensor fabricated by microneedle patch for multiplexed detection of health-related biomarkers. Adv. Sci. 2021, 8, 2103030.
111. Wang, Z.; Li, H.; Wang, J.; et al. Transdermal colorimetric patch for hyperglycemia sensing in diabetic mice. Biomaterials 2020, 237, 119782.
112. Huang, W.; Yang, Y.; Xu, Y.; Xiao, F.; Wang, L. Sweat wearable sensor based on confined pt nanoparticles in 2d conductive metal–organic frameworks for continuous glucose monitoring. Adv. Sci. 2025, 12, e07212.
113. Shu, Y.; Shang, Z.; Su, T.; et al. A highly flexible Ni–Co MOF nanosheet coated Au/PDMS film based wearable electrochemical sensor for continuous human sweat glucose monitoring. Analyst 2022, 147, 1440-8.
114. Ming, T.; Lan, T.; Yu, M.; et al. A novel electrochemical microneedle sensor for highly sensitive real time monitoring of glucose. Microchem. J. 2024, 207, 112021.
115. Bocina, E.; Cocuzza, C.; Vincenzi, C.; Fino, D.; Cauda, V.; Piumetti, M. Enzyme-based biosensors: emerging tools for advanced biomedical applications. Talanta 2026, 300, 129241.
116. Elgrishi, N.; Rountree, K. J.; Mccarthy, B. D.; Rountree, E. S.; Eisenhart, T. T.; Dempsey, J. L. A practical beginner’s guide to cyclic voltammetry. J. Chem. Educ. 2017, 95, 197-206.
117. Semenova, D.; Zubov, A.; Silina, Y. E.; et al. Mechanistic modeling of cyclic voltammetry: a helpful tool for understanding biosensor principles and supporting design optimization. Sens. Actuat. B. Chem. 2018, 259, 945-55.
118. Lee, G.; Park, J.; Chang, Y. W.; Cho, S.; Kang, M.; Pyun, J. Chronoamperometry-based redox cycling for application to immunoassays. ACS. Sens. 2018, 3, 106-12.
119. Baluta, S.; Meloni, F.; Halicka, K.; et al. Differential pulse voltammetry and chronoamperometry as analytical tools for epinephrine detection using a tyrosinase-based electrochemical biosensor. RSC. Adv. 2022, 12, 25342-53.
120. Moreno-Guzmán, M.; Ojeda, I.; Villalonga, R.; González-cortés, A.; Yáñez-Sedeño, P.; Pingarrón, J. M. Ultrasensitive detection of adrenocorticotropin hormone (ACTH) using disposable phenylboronic-modified electrochemical immunosensors. Biosens. Bioelectron. 2012, 35, 82-6.
121. Beitollahi, H.; Tajik, S.; Nejad, F. G. Utilization of MoS2 nanosheets/MnO2 nanorods-based electrochemical sensor for 4-aminophenol determination in the presence of acetaminophen. J. Environ. Chem. Eng. 2025, 13, 117113.
122. Lazanas, A. C.; Prodromidis, M. I. Electrochemical impedance spectroscopy - a tutorial. ACS. Meas. Sci. Au. 2023, 3, 162-93.
123. Srivastava, M.; Nirala, N. R.; Srivastava, S. K.; Prakash, R. A comparative study of aptasensor vs immunosensor for label-free PSA cancer detection on GQDs-AuNRs modified screen-printed electrodes. Sci. Rep. 2018, 8, 1923.
124. Zheng, L.; Zhu, D.; Xiao, Y.; Zheng, X.; Chen, P. Microneedle coupled epidermal sensor for multiplexed electrochemical detection of kidney disease biomarkers. Biosens. Bioelectron. 2023, 237, 115506.
125. Kumar, P.; Jaiwal, R.; Pundir, C. An improved amperometric creatinine biosensor based on nanoparticles of creatininase, creatinase and sarcosine oxidase. Anal. Biochem. 2017, 537, 41-9.
126. Paul, A.; Srivastava, D. N. Amperometric glucose sensing at nanomolar level using MOF-encapsulated TiO2 platform. ACS. Omega. 2018, 3, 14634-40.
127. Wang, Q. Molinero-fernandez, Á.; Wei, Q.; et al. Intradermal lactate monitoring based on a microneedle sensor patch for enhanced in vivo accuracy. ACS. Sens. 2024, 9, 3115-25.
128. Li, F.; Feng, J.; Gao, Z.; et al. Facile synthesis of Cu2O@TiO2-PtCu nanocomposites as a signal amplification strategy for the insulin detection. ACS. Appl. Mater. Interfaces. 2019, 11, 8945-53.
129. Jamalipour Soufi, G.; Iravani, S.; Varma, R. S. Molecularly imprinted polymers for the detection of viruses: challenges and opportunities. Analyst 2021, 146, 3087-100.
130. Haji-hashemi, H.; Bahadorikhalili, S.; Prieto-simón, B. Advancing noninvasive therapeutic drug monitoring via a 3D microstructured aptasensing platform. ACS. Omega. 2025, 10, 35689-97.
131. Wu, Y.; Tehrani, F.; Teymourian, H.; et al. Microneedle aptamer-based sensors for continuous, real-time therapeutic drug monitoring. Anal. Chem. 2022, 94, 8335-45.
132. Kai, H.; Kumatani, A. A porous microneedle electrochemical glucose sensor fabricated on a scaffold of a polymer monolith. J. Phys. Energy. 2021, 3, 024006.
133. Yeasmin, S.; Ullah, A.; Wu, B.; Zhang, X.; Cheng, L. Enzyme-mimics for sensitive and selective steroid metabolite detection. ACS. Appl. Mater. Interfaces. 2023, acsami.2c21980.
134. Tan, F.; Zhai, M.; Meng, X.; Wang, Y.; Zhao, H.; Wang, X. Hybrid peptide-molecularly imprinted polymer interface for electrochemical detection of vancomycin in complex matrices. Biosens. Bioelectron. 2021, 184, 113220.
135. Kilic, N. M.; Singh, S.; Keles, G.; Cinti, S.; Kurbanoglu, S.; Odaci, D. Novel approaches to enzyme-based electrochemical nanobiosensors. Biosensors 2023, 13, 622.
136. Kumar, H. Neelam. Enzyme-based electrochemical biosensors for food safety: a review. Nanobiosens. Dis. Diagn. 2016, 29.
137. Wang, H.; Liu, C.; Feng, T.; et al. Enzyme-based electrochemical sensing systems for on-site detection: recent progress and prospects. Small 2025, 21, e07926.
138. Kozitsina, A.; Svalova, T.; Malysheva, N.; Okhokhonin, A.; Vidrevich, M.; Brainina, K. Sensors based on bio and biomimetic receptors in medical diagnostic, environment, and food analysis. Biosensors 2018, 8, 35.
139. Dzyadevych, S.; Arkhypova, V.; Soldatkin, A.; El’skaya, A.; Martelet, C.; Jaffrezic-renault, N. Amperometric enzyme biosensors: past, present and future. IRBM 2008, 29, 171-80.
140. Jarnda, K. V.; Wang, D. Qurrat-Ul-Ain; et al. Recent advances in electrochemical non-enzymatic glucose sensor for the detection of glucose in tears and saliva: a review. Sens. Actuat. A. Phys. 2023, 363, 114778.
141. Hemalatha, J.; Senthamil, C.; Sakthivel, C.; Nivetha, A.; Umashankar, J.; Prabha, I. Efficient transition metal nanozymes as the alternate for natural enzymes in food analysis and environmental remediation. J. Environ. Chem. Eng. 2024, 12, 112575.
142. He, L.; Ma, X.; Li, Y.; et al. A novel self-powered sensor based on Ni(OH)2/Fe2O3 photoanode for glucose detection by converting solar energy into electricity. J. Alloys. Compd. 2022, 907, 164132.
143. Zhou, F.; Zhao, H.; Chen, K.; Cao, S.; Shi, Z.; Lan, M. Flexible electrochemical sensor with Fe/Co bimetallic oxides for sensitive analysis of glucose in human tears. Anal. Chim. Acta. 2023, 1243, 340781.
144. Wang, L.; Meng, T.; Zhao, D.; et al. An enzyme-free electrochemical biosensor based on well monodisperse Au nanorods for ultra-sensitive detection of telomerase activity. Biosens. Bioelectron. 2020, 148, 111834.
145. Naik, K. K.; Gangan, A.; Chakraborty, B.; Rout, C. S. Superior non-enzymatic glucose sensing properties of Ag-/Au-NiCo2O4 nanosheets with insight from electronic structure simulations. Analyst 2018, 143, 571-9.
146. Ghosh, R.; Li, X.; Yates, M. Z. Nonenzymatic glucose sensor using bimetallic catalysts. ACS. Appl. Mater. Interfaces. 2023, 16, 17-29.
147. Zhu, H.; Shi, F.; Peng, M.; et al. Non-enzymatic electrochemical glucose sensors based on metal oxides and sulfides: recent progress and perspectives. Chemosensors 2025, 13, 19.
148. Verma, S.; Pandey, C. M.; Kumar, D. Non-enzymatic electrochemical biosensor based on MgO@rGO-MoS2 nanohybrid for phenolic compounds detection. Appl. Organomet. Chem. 2023, 38, e7325.
149. Kumari, D.; Prajapati, M.; Ravi Kant, C. Highly efficient non-enzymatic electrochemical glucose biosensor based on copper metal organic framework coated on graphite sheet. ECS. J. Solid. State. Sci. Technol. 2024, 13, 047007.
150. Liu, X., Li, W., Xu, X., Zhou, J., Nie, Z. Electrochemical aptamer sensor for small molecule assays. In Chemical Genomics and Proteomics; Methods in Molecular Biology, Vol. 800; Humana Press, 2011; pp 119-32.
151. Feng, X.; Ju, Y.; Dou, W.; et al. Ferrocene-labelled electroactive aptamer-based sensors (aptasensors) for glycated haemoglobin. Molecules 2021, 26, 7077.
152. Jiang, L.; Liu, N.; Li, D.; et al. A structure-switching electrochemical aptamer sensor for mercury ions based on an ordered assembled gold nanorods-modified electrode. Solid. State. Sci. 2024, 154, 107582.
153. Dalirirad, S.; Han, D.; Steckl, A. J. Aptamer-based lateral flow biosensor for rapid detection of salivary cortisol. ACS. Omega. 2020, 5, 32890-8.
154. Aliakbarinodehi, N.; Jolly, P.; Bhalla, N.; et al. Aptamer-based field-effect biosensor for tenofovir detection. Sci. Rep. 2017, 7, 44409.
155. Chakraborty, M.; Bera, K. K.; Mandal, M.; et al. Phase-dependent electrocatalytic activities of Pt-anchored rutile, anatase and mixed anatase-rutile TiO2 nano-composites for methanol oxidation in alkali. Solid. State. Sci. 2022, 129, 106903.
156. Luo, Y.; Wu, D.; Li, Z.; et al. Plasma functionalized MoSe2 for efficient nonenzymatic sensing of hydrogen peroxide in ultra-wide pH range. SmartMat 2022, 3, 491-502.
157. Yoo, H.; Jo, H.; Oh, S. S. Detection and beyond: challenges and advances in aptamer-based biosensors. Mater. Adv. 2020, 1, 2663-87.
158. Mollarasouli, F.; Kurbanoglu, S.; Ozkan, S. A. The role of electrochemical immunosensors in clinical analysis. Biosensors 2019, 9, 86.
159. Yang, Y.; Yan, Q.; Liu, Q.; et al. An ultrasensitive sandwich-type electrochemical immunosensor based on the signal amplification strategy of echinoidea-shaped Au@Ag-Cu2O nanoparticles for prostate specific antigen detection. Biosens. Bioelectron. 2018, 99, 450-7.
160. Mpofu, K.; Chauke, S.; Thwala, L.; Mthunzi-kufa, P. Aptamers and antibodies in optical biosensing. Discov. Chem. 2025, 2, 23.
161. Ayerdurai, V.; Cieplak, M.; Kutner, W. Molecularly imprinted polymer-based electrochemical sensors for food contaminants determination. TrAC. -. Trends. Anal. Chem. 2023, 158, 116830.
162. Babamiri, B.; Sadri, R.; Farrokhnia, M.; et al. Molecularly imprinted polymer biosensor based on nitrogen-doped electrochemically exfoliated graphene/Ti3 CNTX MXene nanocomposite for metabolites detection. ACS. Appl. Mater. Interfaces. 2024, 16, 27714-27.
163. Kaur, S.; Singla, P.; Dann, A. J.; et al. Sensitive electrochemical and thermal detection of human noroviruses using molecularly imprinted polymer nanoparticles generated against a viral target. ACS. Appl. Mater. Interfaces. 2024, 16, 51397-410.
164. Tchekwagep, P. M. S.; Crapnell, R. D.; Banks, C. E.; et al. A critical review on the use of molecular imprinting for trace heavy metal and micropollutant detection. Chemosensors 2022, 10, 296.
165. Wang, L.; Pagett, M.; Zhang, W. Molecularly imprinted polymer (MIP) based electrochemical sensors and their recent advances in health applications. Sens. Actuat. Rep. 2023, 5, 100153.
166. Cui, R.; Wang, X.; Zhang, G.; Wang, C. Simultaneous determination of dopamine, ascorbic acid, and uric acid using helical carbon nanotubes modified electrode. Sens. Actuat. B. Chem. 2012, 161, 1139-43.
168. Shao, Y.; Wang, J.; Wu, H.; Liu, J.; Aksay, I.; Lin, Y. Graphene based electrochemical sensors and biosensors: a review. Electroanalysis 2010, 22, 1027-36.
169. Latif, U.; Dickert, F. L.; Blach, R. G.; et al. Biocompatible membranes and coatings for glucose sensor. J. Chem. Soc. Pakistan. 2013, 35. , 17-22. https://jcsp.org.pk/PublishedVersion/2d7da3ed-1060-463f-8c9d-8e4bc7ffad47Manuscript%20no%204,%201st%20Gally%20proof%20of%209099%20_UsmanLATIF_.pdf (accessed 2026-05-11).
170. Lubin, A. A.; Plaxco, K. W. Folding-based electrochemical biosensors: the case for responsive nucleic acid architectures. Acc. Chem. Res. 2010, 43, 496-505.
171. Ogurtsova, K.; Da Rocha Fernandes, J.; Huang, Y.; et al. IDF Diabetes Atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes. Res. Clin. Pract. 2017, 128, 40-50.
172. Aschner, P.; Karuranga, S.; James, S.; et al. The International Diabetes Federation’s guide for diabetes epidemiological studies. Diabetes. Res. Clin. Pract. 2021, 172, 108630.
173. Huang, X.; Liang, B.; Huang, S.; et al. Integrated electronic/fluidic microneedle system for glucose sensing and insulin delivery. Theranostics 2024, 14, 1662-82.
174. Mohamad Nor, N.; Ridhuan, N. S.; Abdul Razak, K. Progress of enzymatic and non-enzymatic electrochemical glucose biosensor based on nanomaterial-modified electrode. Biosensors 2022, 12, 1136.
175. Furukawa, H.; Cordova, K. E.; O’keeffe, M.; Yaghi, O. M. The chemistry and applications of metal-organic frameworks. Science 2013, 341, 1230444.
176. Liu, Y.; Dong, Y.; Guo, C. X.; Cui, Z.; Zheng, L.; Li, C. M. Protein-directed in situ synthesis of gold nanoparticles on reduced graphene oxide modified electrode for nonenzymatic glucose sensing. Electroanalysis 2012, 24, 2348-53.
177. Dettmer, M.; Holthaus, C. V.; Fuller, B. M. The impact of serial lactate monitoring on emergency department resuscitation interventions and clinical outcomes in severe sepsis and septic shock: an observational cohort study. Shock 2015, 43, 55-61.
178. Chertoff, J.; Chisum, M.; Garcia, B.; Lascano, J. Lactate kinetics in sepsis and septic shock: a review of the literature and rationale for further research. J. Intensive. Care. 2015, 3, 39.
179. Valvona, C. J.; Fillmore, H. L.; Nunn, P. B.; Pilkington, G. J. The regulation and function of lactate dehydrogenase a: therapeutic potential in brain tumor. Brain. Pathol. 2015, 26, 3-17.
180. Semenza, G. L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664-71.
181. Hui, S.; Ghergurovich, J. M.; Morscher, R. J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115-8.
182. Freeman, D. M. E.; Ming, D. K.; Wilson, R.; et al. Continuous measurement of lactate concentration in human subjects through direct electron transfer from enzymes to microneedle electrodes. ACS. Sens. 2023, 8, 1639-47.
183. Pundir, C.; Yadav, S.; Kumar, A. Creatinine sensors. TrAC. -. Trends. Anal. Chem. 2013, 50, 42-52.
184. Kashani, K.; Rosner, M. H.; Ostermann, M. Creatinine: from physiology to clinical application. Eur. J. Intern. Med. 2020, 72, 9-14.
185. Gao, B.; Li, Y.; Zhang, Z. Preparation and recognition performance of creatinine-imprinted material prepared with novel surface-imprinting technique. J. Chromatogr. B. 2010, 878, 2077-86.
186. Lakshmi, D.; Prasad, B. B.; Sharma, P. S. Creatinine sensor based on a molecularly imprinted polymer-modified hanging mercury drop electrode. Talanta 2006, 70, 272-80.
187. Zinchenko, O.; Marchenko, S.; Sergeyeva, T.; et al. Application of creatinine-sensitive biosensor for hemodialysis control. Biosens. Bioelectron. 2012, 35, 466-9.
188. Saddique, Z.; Faheem, M.; Habib, A.; Ulhasan, I.; Mujahid, A.; Afzal, A. Electrochemical creatinine (bio)sensors for point-of-care diagnosis of renal malfunction and chronic kidney disorders. Diagnostics 2023, 13, 1737.
189. Mohabbati-kalejahi, E.; Azimirad, V.; Bahrami, M.; Ganbari, A. A review on creatinine measurement techniques. Talanta 2012, 97, 1-8.
190. Nieh, C. H.; Tsujimura, S.; Shirai, O.; Kano, K. Amperometric biosensor based on reductive H2O2 detection using pentacyanoferrate-bound polymer for creatinine determination. Anal. Chim. Acta. 2013, 767, 128-33.
191. Dasgupta, P.; Kumar, V.; Krishnaswamy, P. R.; Bhat, N. Serum creatinine electrochemical biosensor on printed electrodes using monoenzymatic pathway to 1-methylhydantoin detection. ACS. Omega. 2020, 5, 22459-64.
192. Jha, V.; Garcia-garcia, G.; Iseki, K.; et al. Chronic kidney disease: global dimension and perspectives. The. Lancet. 2013, 382, 260-72.
193. Romagnani, P.; Remuzzi, G.; Glassock, R.; et al. Chronic kidney disease. Nat. Rev. Dis. Primers. 2017, 3, 17088.
194. Schiffrin, E. L.; Lipman, M. L.; Mann, J. F. Chronic kidney disease: effects on the cardiovascular system. Circulation 2007, 116, 85-97.
195. Walker, V. Ammonia toxicity and its prevention in inherited defects of the urea cycle. Diabetes. Obes. Metab. 2009, 11, 823-35.
196. Foschi, F. G. Urea cycle disorders: a case report of a successful treatment with liver transplant and a literature review. World. J. Gastroenterol. 2015, 21, 4063.
197. Rüfenacht, V.; Häberle, J. Mini-review: challenges in newborn screening for urea cycle disorders. IJNS. 2015, 1, 27-35.
198. Prissanaroon-ouajai, W.; Sirivat, A.; Pigram, P. J.; Brack, N. Potentiometric urea biosensor based on a urease-immobilized polypyrrole. Macromol. Symp. 2015, 354, 334-9.
199. Dervisevic, M.; Jara Fornerod, M. J.; Harberts, J.; Zangabad, P. S.; Voelcker, N. H. Wearable microneedle patch for transdermal electrochemical monitoring of urea in interstitial fluid. ACS. Sens. 2024, 9, 932-41.
200. Djuric, Z.; Bird, C. E.; Furumoto-dawson, A.; et al. Biomarkers of psychological stress in health disparities research. Open. Biomark. J. 2008, 1, 7-19.
201. Kelly, J. J.; Mangos, G.; Williamson, P. M.; Whitworth, J. A. Cortisol and hypertension. Clin. Exp. Pharmacol. Physiol. 1998, 25, S51-6.
202. Iqbal, T.; Simpkin, A. J.; Roshan, D.; et al. Stress monitoring using wearable sensors: a pilot study and stress-predict dataset. Sensors 2022, 22, 8135.
203. Steckl, A. J.; Ray, P. Stress biomarkers in biological fluids and their point-of-use detection. ACS. Sens. 2018, 3, 2025-44.
204. Lamichhane, H. B.; Henares, T. G.; Hackett, M. J.; Arrigan, D. W. M. Structural changes in insulin at a soft electrochemical interface. Anal. Chem. 2021, 93, 9094-102.
205. Soffe, R.; Nock, V.; Chase, J. G. Towards point-of-care insulin detection. ACS. Sens. 2018, 4, 3-19.
206. Schirhagl, R.; Latif, U.; Podlipna, D.; Blumenstock, H.; Dickert, F. L. Natural and biomimetic materials for the detection of insulin. Anal. Chem. 2012, 84, 3908-13.
207. Reaven, G. M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595-607.
208. Monteiro, J. F.; Hahn, S. R.; Gonçalves, J.; Fresco, P. Vancomycin therapeutic drug monitoring and population pharmacokinetic models in special patient subpopulations. Pharmacol. Res. Perspect. 2018, 6, e00420.
209. Gaber, A. M.; Emara, H. M.; Allam, N. K. Electrochemical biosensors for vancomycin monitoring in blood: advances, strategies, and future perspectives. RSC. Adv. 2025, 15, 41418-31.
210. Zhang, T.; Yi, J.; Cheng, H.; et al. Revised therapeutic window for vancomycin in pediatric patients: evidence from a retrospective therapeutic drug monitoring study. BMC. Pharmacol. Toxicol. 2025, 26, 192.
211. Yun, J. H.; Chang, E.; Bae, S.; et al. Risk factors for vancomycin treatment failure in heterogeneous vancomycin-intermediate Staphylococcus aureus bacteremia. Microbiol. Spectr. 2024, 12, e00333-24.
212. Wang, L.; Wang, Y.; Yang, C.; Jiang, J.; Wang, H.; Wu, M. Multiple linear regression model was constructed based on the influencing factors of vancomycin trough concentration. Dose-Response 2025, 23, 15593258251313646.
213. Lin, L.; Lian, H.; Sun, X.; Yu, Y.; Liu, B. An L-dopa electrochemical sensor based on a graphene doped molecularly imprinted chitosan film. Anal. Methods. 2015, 7, 1387-94.
215. Puris, E.; Gynther, M.; Auriola, S.; Huttunen, K. M. L-Type amino acid transporter 1 as a target for drug delivery. Pharm. Res. 2020, 37, 88.
216. Kiss, T.; Katona, G.; Mérai, L.; et al. Development of a hydrophobicity-controlled delivery system containing levodopa methyl ester hydrochloride loaded into a mesoporous silica. Pharmaceutics 2021, 13, 1039.
217. Senek, M.; Nyholm, D.; Nielsen, E. I. Population pharmacokinetics of levodopa/carbidopa microtablets in healthy subjects and Parkinson’s disease patients. Eur. J. Clin. Pharmacol. 2018, 74, 1299-307.
219. Jankovic, J.; Tan, E. K. Parkinson’s disease: etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry. 2020, 91, 795-808.
220. Tan, A. H.; Chuah, K. H.; Beh, Y. Y.; Schee, J. P.; Mahadeva, S.; Lim, S. Gastrointestinal dysfunction in Parkinson’s disease: neuro-gastroenterology perspectives on a multifaceted problem. JMD. 2023, 16, 138-51.
222. Goud, K. Y.; Moonla, C.; Mishra, R. K.; et al. Wearable electrochemical microneedle sensor for continuous monitoring of levodopa: toward parkinson management. ACS. Sens. 2019, 4, 2196-204.
223. Fang, L.; Ren, H.; Mao, X.; et al. Differential amperometric microneedle biosensor for wearable levodopa monitoring of Parkinson’s disease. Biosensors 2022, 12, 102.
224. Manyanga, V.; Elkady, E.; Hoogmartens, J.; Adams, E. Improved reversed phase liquid chromatographic method with pulsed electrochemical detection for tobramycin in bulk and pharmaceutical formulation. J. Pharm. Anal. 2013, 3, 161-7.
225. Arsand, J. B.; Jank, L.; Martins, M. T.; et al. Determination of aminoglycoside residues in milk and muscle based on a simple and fast extraction procedure followed by liquid chromatography coupled to tandem mass spectrometry and time of flight mass spectrometry. Talanta 2016, 154, 38-45.
226. Lin, S.; Cheng, X.; Zhu, J.; et al. Wearable microneedle-based electrochemical aptamer biosensing for precision dosing of drugs with narrow therapeutic windows. Sci. Adv. 2022, 8, eabq4539.
227. Wei, Y.; Chen, W.; Ma, Y.; et al. Integrated individually addressable microneedle arrays for robust glucose monitoring and on-demand insulin releasing. Microsyst. Nanoeng. 2025, 11, 211.
228. Rivnay, J.; Inal, S.; Salleo, A.; Owens, R. M.; Berggren, M.; Malliaras, G. G. Organic electrochemical transistors. Nat. Rev. Mater. 2018, 3, 17086.
229. Cui, F.; Yue, Y.; Zhang, Y.; Zhang, Z.; Zhou, H. S. Advancing biosensors with machine learning. ACS. Sens. 2020, 5, 3346-64.
230. Wu, M.; Li, L.; Yu, R.; et al. Tailored diffusion limiting membrane for microneedle glucose sensors with wide linear range. Talanta 2024, 273, 125933.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.